
Filed Pursuant to Rule 253(g)(3)
File No. 024-11602
Offering Circular dated December 13, 2021
RespireRx Pharmaceuticals Inc.
126 Valley Road, Suite C
Glen Rock, New Jersey 07452
(201) 444-4947
Maximum Offering Amount: $7,500,000
This is a public offering (this “Offering”) of securities of RespireRx Pharmaceuticals Inc., a Delaware corporation (“RespireRx” and together with RespireRx’s wholly owned subsidiary, Pier Pharmaceuticals, Inc. (“Pier”), the “Company,” “we,” or “our,” unless the context indicates otherwise). We are offering a maximum of 375,000,000 shares (the “Shares”) of Common Stock, par value $0.001 per share (“Common Stock”), at an offering price of $0.02 per Share, up to a maximum of $7,500,000 (“Maximum Offering”), on a “best efforts” basis. This Offering will expire on the first to occur of (a) the sale of all 375,000,000 Shares offered hereby, (b) October 31, 2023, or (c) when the Company’s board of directors elects to terminate the Offering (as applicable, the “Termination Date”). There is no escrow established for this Offering. We will receive subscriptions through prospective investors’ submissions of subscription agreements (“Subscription Agreements”), which will include investor qualification questionnaires. We will provide instructions for payment/funding of the investment only to those investors from whom we have received completed Subscription Agreements, and only after review and acceptance of such Subscription Agreements. The Shares will be sold for cash or may be issued as repayment of accounts payable, accrued expenses, principal on promissory notes, convertible or otherwise, inclusive or exclusive of interest, or other liabilities, all or any of which might occur without notice to subscribers, and would only occur pursuant to Subscription Agreements accepted by the Company. Funds received from individuals or entities seeking to invest with respect to which we have not received or accepted Subscription Agreements will be returned to such individuals or entities and any intended investment will be considered null and void ab initio. We will hold closings upon the receipt and acceptance of investors’ Subscription Agreements and receipt of invested funds by the Company. If, on the initial closing date, we have sold less than the Maximum Offering, then we may hold one or more additional closings for additional sales, until the earlier of: (i) the sale of the Maximum Offering or (ii) the Termination Date. There is no aggregate minimum requirement for the Offering to hold a closing, therefore, we reserve the right, subject to applicable securities laws, to begin applying “dollar one” of the proceeds from the Offering in accordance with the “Use of Proceeds” section of this offering circular (this “Offering Circular”) and such other uses as more specifically set forth in this Offering Circular. We expect to commence the sale of the Shares within two days of the date on which the offering statement on Form 1-A (the “Offering Statement”) of which this Offering Circular is a part is qualified by the United States Securities and Exchange Commission (the “SEC”).
The Company’s Common Stock is listed on the OTCQB Venture Market (the “OTCQB”), under the symbol “RSPI.” The last sales price for the Company’s Common Stock on December 10, 2021 was $ 0.016. For further information, see “Plan of Distribution” of this Offering Circular.
The offering price of $0.02 per share was determined by management in order to attract investors in this Offering and is based on a discount to the trading price of our Common Stock on the OTCQB over the past six months, taking into account trading volume, price range, volume weighted average pricing and other factors. The price range also reflects prices at which we believe we can sell such shares in a timely manner, and is not based on book value, assets, earnings or any other recognizable standard of value.
The narrative disclosure in this Offering Circular follows the Form S-1 format pursuant to Part II(A)(1)(ii) of Form 1-A.
Investing in our Shares involves a high degree of risk. See “Risk Factors” beginning on page 10 for a discussion of certain risks that you should consider in connection with an investment in our Common Stock.
THE UNITED STATES SECURITIES AND EXCHANGE COMMISSION DOES NOT PASS UPON THE MERITS OF OR GIVE ITS APPROVAL TO ANY SECURITIES OFFERED OR THE TERMS OF THE OFFERING, NOR DOES IT PASS UPON THE ACCURACY OR COMPLETENESS OF ANY OFFERING CIRCULAR OR OTHER SOLICITATION MATERIALS. THESE SECURITIES ARE OFFERED PURSUANT TO AN EXEMPTION FROM REGISTRATION WITH THE COMMISSION; HOWEVER, THE COMMISSION HAS NOT MADE AN INDEPENDENT DETERMINATION THAT THE SECURITIES OFFERED ARE EXEMPT FROM REGISTRATION.
GENERALLY, NO SALE MAY BE MADE TO YOU IN THIS OFFERING IF THE AGGREGATE PURCHASE PRICE YOU PAY IS MORE THAN 10% OF THE GREATER OF YOUR ANNUAL INCOME OR NET WORTH. DIFFERENT RULES APPLY TO ACCREDITED INVESTORS AND NON-NATURAL PERONS. BEFORE MAKING ANY REPRESENTATION THAT YOUR INVESTMENT DOES NOT EXCEED APPLICABLE THRESHOLDS, WE ENCOURAGE YOU TO REVIEW RULE 251(d)(2)(i)(C) OF REGULATION A. FOR GENERAL INFORMATION ON INVESTING, WE ENCOURAGE YOU TO REFER TO WWW.INVESTOR.GOV.
| Price to Public | Commissions | Proceeds to the Company | ||||||||||
| Per Share | $ | 0.02 | (1) | $ | 0.0014 | (2) | $ | 0.0186 | ||||
| Maximum Offering | $ | 7,500,000 | $ | 525,000 | $ | 6,975,000 | (3) | |||||
(1) We are offering Shares at an offering price of $0.02 per share.
(2) The Placement Agent is entitled to a fee of 7% of the gross proceeds for sales to institutional accredited investors originated by the Placement Agent and 4% of the gross proceeds for sales to institutional accredited investors referred to the Placement Agent by the Company. We have also agreed to issue warrants to purchase shares of our Common Stock to the Placement Agent and to reimburse the Placement Agent and any co-agents for certain expenses. See “Plan of Distribution” on page 21 for additional information regarding total placement agent compensation.
(3) Net proceeds to the Company assuming the Maximum Offering (before offering expenses).
THIS OFFERING CIRCULAR IS NOT AN OFFER TO SELL, NOR SOLICITING AN OFFER TO BUY, ANY SHARES OF OUR COMMON STOCK IN ANY STATE OR OTHER JURISDICTION IN WHICH SUCH SALE IS PROHIBITED.
The date of this Offering Circular is December 13, 2021
Table
of Contents
| 3 |
As used in this Offering Circular, all references to “RespireRx,” the “Company,” “we,” “our,” “Shares” “capital stock,” “Common Stock,” “Series B Preferred Stock,” “Preferred Stock” or “stockholders,” applies only to RespireRx Pharmaceuticals Inc. As used in this Offering Circular, the terms “consolidated we,” “consolidated our” or words of like import mean RespireRx Pharmaceuticals Inc. and its direct wholly-owned subsidiary, Pier Pharmaceutical, Inc. Notwithstanding the foregoing, references to the “Company,” “we,” “our” and similar terms that appear in the consolidated financial statements in our annual reports on Form 10-K and in our condensed consolidated financial statements in our quarterly reports on Form 10-Q, refer to RespireRx Pharmaceuticals Inc. and its direct wholly-owned subsidiary Pier Pharmaceuticals, Inc. All references in this Offering Circular to “years” and “fiscal years” means the twelve-month period ended December 31st, unless the context indicates otherwise.
Circumstances may change so as to alter the information presented herein at a later date. This material will be updated by Amendment to this document and by means of press releases and other communications to stockholders.
Use of Market and Industry Data
This Offering Circular includes market and industry data that we have obtained from third-party sources, including industry publications, as well as industry data prepared by our management on the basis of its knowledge of and experience in the industries in which we operate (including our management’s estimates and assumptions relating to such industries based on that knowledge). Management has developed its knowledge of such industries through its experience, participation in and observation of these industries. While our management believes the third-party sources referred to in this Offering Circular are reliable, neither we nor our management have independently verified any of the data from such sources referred to in this Offering Circular or ascertained the underlying economic assumptions relied upon by such sources. Furthermore, internally prepared and third-party market prospective information, in particular, are estimates only and there will usually be differences between the prospective and actual results because events and circumstances frequently do not occur as expected, and those differences may be material. Also, references in this Offering Circular to any publications, reports, surveys or articles prepared by third parties should not be construed as depicting the complete findings of the entire publication, report, survey or article. The information in any such publication, report, survey or article is not incorporated by reference in this Offering Circular.
| 4 |
Cautionary Statements Regarding Forward-Looking Statements
This Offering Circular contains certain forward-looking statements within the meaning of Section 27A of the Securities Act of 1933, as amended (the “Securities Act”) and Section 21E of the Securities Exchange Act of 1934, as amended (the “Exchange Act”), and the Company intends that such forward-looking statements be subject to the safe harbor created thereby. These might include statements regarding the Company’s future plans, targets, estimates, assumptions, financial position, business strategy and other plans and objectives for future operations, and assumptions and predictions about research and development efforts, including, but not limited to, preclinical and clinical research design, execution, timing, costs and results, future product demand, supply, manufacturing, costs, marketing and pricing factors.
In some cases, forward-looking statements may be identified by words including “assumes,” “could,” “ongoing,” “potential,” “predicts,” “projects,” “should,” “will,” “would,” “anticipates,” “believes,” “intends,” “estimates,” “expects,” “plans,” “contemplates,” “targets,” “continues,” “budgets,” “may,” or the negative of these terms or other comparable terminology, although not all forward-looking statements contain these words, and such statements may include, but are not limited to, statements regarding (i) future research plans, expenditures and results, (ii) potential collaborative arrangements, (iii) the potential utility of the Company’s products candidates, (iv) reorganization plans, and (v) the need for, and availability of, additional financing. Forward-looking statements are based on information available at the time the statements are made and involve known and unknown risks, uncertainties and other factors that may cause our results, levels of activity, performance or achievements to be materially different from the information expressed or implied by the forward-looking statements in this Offering Circular.
These factors include but are not limited to, regulatory policies or changes thereto, available cash, research and development results, issuance of patents, competition from other similar businesses, interest of third parties in collaborations with us, and market and general economic factors, and other risk factors disclosed in “Item 1A. Risk Factors” in the Company’s Annual Report on Form 10-K for the fiscal year ended December 31, 2020, as filed with the SEC on April 15, 2021 (the “2020 Form 10-K”).
You should read these risk factors and the other cautionary statements made in the Company’s filings as being applicable to all related forward-looking statements wherever they appear in this Offering Circular. We cannot assure you that the forward-looking statements in this Offering Circular will prove to be accurate and therefore prospective investors are encouraged not to place undue reliance on forward-looking statements. You should read this Offering Circular completely. Other than as required by law, we undertake no obligation to update or revise these forward-looking statements, even though our situation may change in the future.
We caution investors not to place undue reliance on any forward-looking statement that speaks only as of the date made and to recognize that forward-looking statements are predictions of future results, which may not occur as anticipated. Actual results could differ materially from those anticipated in the forward-looking statements and from historical results, due to the risks and uncertainties described in the 2020 Form 10-K and in this Offering Circular, as well as others that we may consider immaterial or do not anticipate at this time. These forward-looking statements are based on assumptions regarding the Company’s business and technology, which involve judgments with respect to, among other things, future scientific, economic, regulatory and competitive conditions, collaborations with third parties, and future business decisions, all of which are difficult or impossible to predict accurately and many of which are beyond the Company’s control. Although we believe that the expectations reflected in our forward-looking statements are reasonable, we do not know whether our expectations will prove correct. Our expectations reflected in our forward-looking statements can be affected by inaccurate assumptions that we might make or by known or unknown risks and uncertainties, including those described in the 2020 Form 10-K and in this Offering Circular. These risks and uncertainties are not exclusive and further information concerning us and our business, including factors that potentially could materially affect our financial results or condition, may emerge from time to time.
For more information about the risks and uncertainties the Company faces, see the section “Risk Factors” in this Offering Circular and “Item 1A. Risk Factors” in our 2020 Form 10-K. Forward-looking statements speak only as of the date they are made. The Company does not undertake and specifically declines any obligation to update any forward-looking statements or to publicly announce the results of any revisions to any statements to reflect new information or future events or developments. We advise investors to consult any further disclosures we may make on related subjects in our annual reports on Form 10-K and other reports that we file with or furnish to the SEC.
| 5 |
This summary highlights information contained elsewhere in this Offering Circular and does not contain all of the information that you should consider in making your investment decision. Before investing in our securities, you should carefully read this entire Offering Circular, including our financial statements and the related notes and the information set forth under the headings “Risk Factors” and “Management’s Discussion and Analysis of Financial Condition and Results of Operations” in each case included elsewhere in this Offering Circular.
Overview
The mission of the Company is to develop innovative and revolutionary treatments to combat disorders caused by disruption of neuronal signaling. We are developing treatment options that address conditions that affect millions of people, but for which there are limited or poor treatment options, including obstructive sleep apnea (“OSA”), attention deficit hyperactivity disorder (“ADHD”), epilepsy, chronic pain, including inflammatory and neuropathic pain, recovery from spinal cord injury (“SCI”), as well as other areas of interest based on results of preclinical and clinical studies to date.
RespireRx is developing a pipeline of new drug products based on our broad patent portfolios across two distinct drug platforms:
| (i) | our pharmaceutical cannabinoids platform (which we refer to as ResolutionRx ) is developing compounds that target the body’s endocannabinoid system, and in particular, the re-purposing of dronabinol, an endocannabinoid CB1 and CB2 receptor agonist, for the treatment of OSA. Dronabinol is already approved by the FDA for other indications. | |
| (ii) | our neuromodulators platform (which we refer to as EndeavourRx) is made up of two programs: (a) our AMPAkines program, which is developing proprietary compounds that are positive allosteric modulators (“PAMs”) of AMPA-type glutamate receptors to promote neuronal function and (b) our GABAkines program, which is developing proprietary compounds that are PAMs of GABAA receptors, and which was recently established pursuant to our entry with the University of Wisconsin-Milwaukee Research Foundation, Inc., an affiliate of the University of Wisconsin-Milwaukee (“UWMRF”), into a patent license agreement (the “UWMRF Patent License Agreement”). |
Recent Developments
We have been assessing the impact of the COVID-19 pandemic on our discovery, research and clinical programs, including impacts on their expected timelines and costs. Because we are not actively pursuing any clinical trials at this time due to insufficient funding, the pandemic has not impacted our clinical program operations significantly; however, if we are able to secure financing or otherwise can proceed with clinical development, these impacts could ultimately be detrimental. On March 18, 2020, July 2, 2020, and January 27, 2021, the U.S. Food and Drug Administration (“FDA”) issued updated industry guidance for conducting clinical trials, in which the FDA emphasized that safety of trial participants is critically important. This guidance may lead to the implementation of additional protocols such as COVID-19 screening procedures, resulting in potential delays and additional costs. The risks, strategic and operational challenges and costs of conducting such trials as a result of the global pandemic have exacerbated an already challenging clinical trial process. See “Risk Factors” for more information regarding the potential impact of the COVID-19 pandemic on our business and operations. We will continue to evaluate the impact of the COVID-19 pandemic on our business.
Our major challenge has been to raise substantial equity or equity-linked financing to support research and development plans for our cannabinoid (ResolutionRx) and neuromodulator (EndeavourRx) business platforms, while minimizing the dilutive effect to pre-existing stockholders. At present, we believe that we are hindered primarily by our public corporate structure, our OTCQB listing, and low market capitalization as a result of our low stock price. See “Risk Factors—Risks related to capital structure—If our common stock is determined to be a “penny stock,” a broker-dealer may find it more difficult to trade our common stock and an investor may find it more difficult to acquire or dispose of our common stock in the secondary market.”
For this reason, the Company has begun to implement an internal restructuring plan through which our two drug platforms, ResolutionRx and EndeavourRx, have been reorganized into separate business divisions and may in the future be organized into subsidiaries of RespireRx. We believe that by creating one or more subsidiaries to further the aims of ResolutionRx and EndeavourRx, it may be possible, through separate finance channels, to optimize the asset values of each.
Risks Associated with Our Business
Our business is subject to many risks, as more fully described in the section titled “Risk Factors” in this Offering Circular. You should read and carefully consider these risks, together with the risks set forth under the section titled “Risk Factors” and all of the other information in this Offering Circular, including the financial statements and the related notes included elsewhere in this Offering Circular, before deciding whether to invest in our securities. If any of the risks discussed in this Offering Circular actually occur, our business, financial condition or operating results could be materially and adversely affected. In particular, such risks include, but are not limited to, the following:
| ● | Our business is subject to risks arising from epidemic diseases, such as the COVID-19 pandemic. | |
| ● | As a result of our current negative net worth, lack of cash and other liquid resources, the magnitude of our liabilities and the difficulties we have historically experienced raising capital, we and our auditors have expressed substantial doubt regarding our ability to continue as a “going concern.” |
| 6 |
| ● | We and our independent registered public accounting firm has identified material weaknesses in our internal financial controls and reporting processes. | |
| ● | Raising additional capital may cause dilution to our stockholders. | |
| ● | Our success, at least in part, will be dependent upon the strength of our intellectual property, including, but not limited to, licensed and owned patents, patent applications, continuations-in-part, provisional patent applications, know-how, trade secrets and other forms of intellectual property. The issuance of patents with relevant claims is subject to varying degrees of uncertainty. Our ability to defend our intellectual property or challenge third party intellectual property infringement claims is expensive, time-consuming and uncertain. If our patent applications do not issue with relevant claims or if we cannot defend our patents, or, as appropriate, challenge interfering patents or actions of third parties, or otherwise maintain our intellectual property, our business and operations will be adversely affected. | |
| ● | Our success is dependent upon our ability to enter into strategic alliances with larger companies in our industry or with companies that have specific expertise. We may not be able to enter into such alliances on terms acceptable to us and our inability to do so would have a material adverse effect on our business. | |
| ● | The markets for our product candidates are highly competitive and are subject to change due to scientific advancements, which could have a material adverse effect on our business, results of operations and financial condition. | |
| ● | One of our product candidates is based, at least in part, on the development of one or more new formulations and the repurposing of an approved drug, the development of which is inherently risky while others of our product candidates have never been approved for marketing by any regulatory bodies and are subject to substantial research and development risks. Concerns about the safety and efficacy of our product candidates could limit our future success. | |
| ● | Clinical trials required for our product candidates are expensive and time-consuming, and their outcomes are highly uncertain. If we are unable to commence our planned clinical trials, or if any of those clinical trials are delayed or yield unfavorable results, we may have to delay application for or may be unable to obtain regulatory approval for the marketing of our product candidates. | |
| ● | Due to our reliance on third parties to conduct clinical trials on our behalf, we are unable to directly control the timing, conduct, expense and quality of our clinical trials, which could adversely affect our clinical data and results and related regulatory approvals. | |
| ● | Our Common Stock is not listed on a national securities exchange and is considered a “penny stock,” with a low market capitalization, all of which makes it more difficult for our stock to trade in the financial markets, for research analysts at securities brokerage firms to write research reports about us, for investment banks to contract with us for services, and ultimately making it difficult for us to obtain necessary capital required to execute our business plan, which could restrict our ability to continue as a going concern and to grow. | |
| ● | Regulatory and legal uncertainties could result in significant costs or otherwise harm our business. | |
| ● | Our directors, executive officers and significant stockholders have substantial control over us and could limit stockholders’ ability to influence the outcome of key transactions, including changes of control. | |
| ● | The Company has the ability to issue series of preferred stock with rights and privileges as established by the Board of Directors and without shareholder consent. This may be used to prevent certain actions, including but not limited to, changes of control. |
Implications of Being a Smaller Reporting Company
We are a “smaller reporting company” as defined in Rule 12b-2 of the Securities Exchange Act of 1934, as amended (the “Exchange Act”), and have elected to take advantage of certain of the scaled disclosure available to smaller reporting companies.
Corporate History
The Company was incorporated in the State of Delaware in 1987 under the name Cortex Pharmaceuticals, Inc. to engage in the discovery, development and commercialization of innovative pharmaceuticals for the treatment of neurological and psychiatric disorders. On December 16, 2015, the Company filed a Certificate of Amendment to its Second Restated Certificate of Incorporation (as amended to date, our “Certificate of Incorporation”) with the Secretary of State of the State of Delaware to change its name from Cortex Pharmaceuticals, Inc. to RespireRx Pharmaceuticals Inc.
| 7 |
In August 2012, the Company acquired Pier Pharmaceuticals, Inc., a Delaware corporation (“Pier”), pursuant to an Agreement and Plan of Merger between the Company and Pier by which Pier merged with and into a subsidiary of the Company, resulting in Pier being a wholly-owned subsidiary of the Company. Pier was a clinical stage biopharmaceutical company developing a pharmacologic treatment for OSA and had been engaged in research and clinical development activities, which are now being conducted by the Company as Pier’s parent.
Our common stock is currently quoted on the OTCQB with the ticker symbol RSPI. RespireRx has never paid dividends on its Common Stock and does not anticipate doing so in the foreseeable future.
Corporate Information
Our corporate mailing address is 126 Valley Road, Suite C, Glen Rock, NJ 07452. Our telephone number is (201) 444-4947, and the contact person for general corporate matters, including investor relations, is Jeff Eliot Margolis, at jmargolis@respirerx.com or (917) 834-7206. Our website is www.respirerx.com, where you will find, among other things, a description of our business, backgrounds of management, presentations, generally in the form of slide decks, press releases and links to our filings with the SEC, which filings are also available at www.sec.gov. The information on our website is not part of this Offering Circular and is not incorporated by reference into, and should not be considered part of, this Offering Circular. Any information about us on LinkedIn, Twitter or other social media platforms should not be considered part of this Offering Circular, nor should any information about us posted by others on blogs, bulletin boards, in chat rooms or in similar media. This Offering is only made via this Offering Circular and our Offering Statement, as amended or supplemented from time to time.
The RespireRx logo and certain trademarks of RespireRx Pharmaceuticals Inc. of or relating to any of its product candidates or program and platform names appearing in this Offering Circular are our property.
| 8 |
| Common Stock We are Offering (“Shares”) | Up to 375,000,000 | |
| Offering Price Per Share (1) | $0.02 | |
| Maximum Offering Amount | $7,500,000 | |
| Common Stock Outstanding Before the Offering(2) | 90,395,596 | |
| Common Stock Outstanding After the Offering Assuming the Maximum Amount is Raised (there is no minimum)(3) |
469,907,943 | |
| Use of Proceeds(4) | The net proceeds of this Offering, after deducting commissions and estimated offering expenses, and assuming the sale of 375,000,000 Shares at $0.02 per share, are expected to be $6,760,000 | |
| We intend to use the net proceeds from this Offering for, among other things, dronabinol research and development, AMPAkines research and development, GABAkines research and development, payment of accounts payable and certain debt, general and administrative expenses, and working capital expenses, all as further explained in “Use of Proceeds” appearing elsewhere in this Offering Circular. | ||
| Risk Factors | See “Risk Factors” on page 10 and other information appearing elsewhere in this Offering Circular as well as in our 2020 Form 10-K and other materials filed and furnished with the SEC. | |
| OTCQB Ticker Symbol | RSPI |
(1) The Shares will be sold for cash or may be issued as repayment of accounts payable, accrued expenses, principal on promissory notes, convertible or otherwise, inclusive or exclusive of interest, or other liabilities, all or any of which might occur without notice to subscribers, and would only occur pursuant to Subscription Agreements accepted by the Company. Only Shares offered for cash will be issued and sold at the first closing.
(2) The number of shares is based on 90,396,596 shares of Common Stock outstanding as of September 30, 2021 and excludes, as of such date (unless otherwise specified):
| ● | 7,111,924 shares issuable upon the exercise of outstanding stock options as of September 30, 2021, | |
| ● | 15,757,542 shares reserved and available for future issuances under our equity plans as of September 30, 2021, | |
| ● | 59,505,140 shares issuable upon the exercise of stock purchase warrants outstanding*, | |
| ● | 40,542,857 shares issuable upon conversion of convertible promissory notes outstanding*, and | |
| ● | 649 shares issuable as “Pier Contingent Shares.” |
* Does not include or account for (a) shares issuable upon conversion of additional interest accrued after September 30, 2021, (b) 5,750,000 shares issuable upon conversion of a convertible promissory note issued on October 7, 2021, or 5,750,000 shares issuable upon exercise of warrants issued on the same date in connection with such convertible promissory note, (c) 4,000,000 shares issued on November 8, 2021 upon partial conversion of $80,000 of a $112,000 convertible note, which reduced the principal amount of such note to $32,000 and reduced the number of shares into which such note is convertible by 4,000,000 shares, (d) the partial cashless exercise on November 8, 2021, of a warrant which reduced the number of warrants by 1,534,042 and resulted in the issuance of 511,347 shares of common stock, or (e) the cashless exercise on November 22, 2021, of a warrant which reduced the number of warrants by 4,300,000 and resulted in the issuance of 1,433,333 shares of common stock.
(3) Excludes shares referenced in footnote (1) and the shares of common stock issuable upon the exercise of warrants issued to the Placement Agent. See “Plan of Distribution.”
(4) See “Use of Proceeds” elsewhere in this Offering Circular for a description of the expected use of proceeds if less than 375,000,000 Shares are sold in this Offering.
| 9 |
The following is only a summary of the risks pertaining to our Company. Investment in our securities involves risks. You should carefully consider the following risk factors in addition to other information contained in this Offering Circular as well as in our 2020 Form 10-K and other materials filed and furnished with the SEC. The occurrence of any of the following risks might cause you to lose all or part of your investment. Some statements in this Offering Circular, including statements in the following risk factors, constitute “forward-looking statements.”
Risks related to our business
We and our independent registered public accounting firm has expressed substantial doubt about our ability to continue as a going concern.
The Company has incurred net losses of $708,682 and $2,045,446 for the three-months and nine-months ended September 30, 2021, respectively, and $4,301,211 for the fiscal year ended December 31, 2020, as well as negative operating cash flows of $800,622 for the nine-months ended September 30, 2021 and $513,001 for the fiscal year ended December 31, 2020. The Company also had a stockholders’ deficiency of $9,424,888 at September 30, 2021 and expects to continue to incur net losses and negative operating cash flows for at least the next few years. As a result, in its audit opinion issued in connection with our consolidated financial statements as of December 31, 2020 and 2019, our independent registered public accounting firm has expressed substantial doubt about our ability to continue as a going concern given our limited working capital, recurring net losses and negative cash flows from operations. The accompanying consolidated financial statements have been prepared on a going concern basis, which contemplates the realization of assets and the satisfaction of liabilities and commitments in the normal course of business. The consolidated financial statements do not include any adjustments relating to the recoverability and classification of recorded asset amounts or amounts of liabilities that might be necessary should we be unable to continue in existence. While we have relied principally in the past on external financing to provide liquidity and capital resources for our operations, we can provide no assurance that cash generated from our operations together with cash received in the future from external financing, if any, will be sufficient to enable us to continue as a going concern.
We and our independent registered public accounting firm has identified material weaknesses in our financial reporting process.
At December 31, 2020, management and our independent registered public accounting firm identified material weaknesses in our internal control over financial reporting. There can be no assurance that we will be able to successfully implement our plans to remediate the material weaknesses in our financial reporting process. Our failure to successfully implement our plans to remediate these material weaknesses could cause us to fail to meet our reporting obligations, to produce timely and reliable financial information, and to effectively prevent fraud. Additionally, such failure, or other weaknesses that we may experience in our financial reporting process or other internal controls, could cause investors to lose confidence in our reported financial information, which could have a negative impact on our financial condition and stock price.
We have a history of net losses; we expect to continue to incur net losses and we may never achieve or maintain profitability.
Since our formation on February 10, 1987 through the end of our most recent fiscal year ended December 31, 2020 and through September 30, 2021, we have generated only minimal operating revenues, primarily from grants for research and development. For the fiscal year ended December 31, 2020, our net loss was $4,301,211 and as of December 31, 2020, we had an accumulated deficit of $170,810,296. For the nine-months ended September 30, 2021, our net loss was $2,045,446 and as of September 30, 2021, we had an accumulated deficit of $173,181,441. We have not generated any revenue from product sales to date, and it is possible that we will never generate revenues from product sales in the future. Even if we do achieve significant revenues from product sales, we expect to continue to incur significant net losses over the next several years. As with other biotechnology companies, it is possible that we will never achieve profitable operations.
We will need additional capital in the near term and the future and, if such capital is not available on terms acceptable to us or available to us at all, we may need to scale back our research and development efforts and may be unable to continue our business operations.
We require additional cash resources for basic operations and will require substantial additional funds to advance our research and development programs and to continue our operations, particularly if we decide to independently conduct later-stage clinical testing and apply for regulatory approval of any of our proposed products, and if we decide to independently undertake the marketing and promotion of our products. Additionally, we may require additional funds in the event that we decide to pursue strategic acquisitions of or licenses for other products or businesses. Based on our operating plan as of December 31, 2020, we estimated that our existing cash resources will not be sufficient to meet our requirements for 2021. We also need additional capital in the near term to fund on-going operations including basic operations. Additional funds may come from the sale of common equity, preferred equity, convertible preferred equity or equity-linked securities, debt, including debt convertible into equity, or may result from agreements with larger pharmaceutical companies that include the license or rights to the technologies and products that we are currently developing, although there is no assurance that we will secure any such funding or other transaction in a timely manner, or at all.
| 10 |
Our cash requirements in the future may differ significantly from our current estimates, depending on a number of factors, including:
| ● | Our ability to raise equity or debt capital, or our ability to obtain in-kind services which may be more difficult during the current pandemic health crisis; | |
| ● | the results of our clinical trials; | |
| ● | the time and costs involved in obtaining regulatory approvals; | |
| ● | the costs associated with the implementation of a corporate restructure | |
| ● | the costs of setting up and operating our own marketing and sales organization; | |
| ● | the ability to obtain funding under contractual and licensing agreements; | |
| ● | the ongoing obligations to make contractual licensed patent maintenance fees, milestone payments and royalty payments; | |
| ● | the costs involved in filing, prosecuting, maintaining and enforcing patents or any litigation by third parties regarding intellectual property; | |
| ● | the costs involved in meeting our contractual obligations including employment agreements; and | |
| ● | our success in entering into collaborative relationships with other parties. |
Common Stock reserve requirements may restrict our ability to raise capital and continue to operate our business.
Common Stock reserve requirements may restrict our ability to raise capital and continue to operate our business. The Company is authorized to issue up to 2 billion (2,000,000,000) shares of Common Stock under its Certificate of Incorporation. As of September 30, 2021, there were 90,396,596 shares of Common Stock issued and outstanding and the Company was required to reserve an aggregate of 206,874,138 shares of its authorized and unissued Common Stock with respect to convertible notes, convertible Series B Preferred Stock, warrants, options granted not yet exercised and shares available for issuance its equity plans, inclusive of incremental contractual reserves in excess of the calculated number of conversion shares and warrant shares. There are 1,702,729,266 authorized, unissued and unreserved shares of Common Stock available after reserving for the incremental contractual reserves of 83,949,700. If we breach the contractual reserve requirements, we will be in default of such contractual obligations which may have material adverse consequences which may make it more difficult to raise additional necessary capital to operate our business. The requirement to reserve an aggregate of 206,874,138 shares as of September 30, 2021 does not include the requirement to reserve 18,125,000 shares with respect to a convertible note and warrant issued on October 7, 2021. Furthermore, the reserve requirement does not reflect reductions therein resulting from: the issuance of 4,000,000 shares of common stock on November 8, 2021 upon the partial conversion of a convertible note (which issuance would reduce the reserve requirements by 12,000,000 shares of common stock), the reduction in the number of shares into which a warrant may be exercised by 1,534,042 share of common stock related to the partial cashless exercise of that warrant (which also would reduce the reserve requirement by 1,534,042 shares of common stock) and the reduction in the number of shares into which a warrant may be exercised by 4,300,000 share of common stock related to the cashless exercise of that warrant (which also would reduce the reserve requirement by 4,300,000 shares of common stock).
Our product opportunities rely on licenses from research institutions and if we lose access to these technologies or applications, our business could be substantially impaired.
Through our acquisition of Pier, we gained access to a pre-existing relationship between Pier and the University of Illinois at Chicago (the “UIC”). Effective in September 2014, the Company entered into a license agreement with the UIC (the “UIC License Agreement”), which gave the Company certain exclusive rights with respect to certain patents and patent applications in the United States and other countries claiming the use of dronabinol and other cannabinoids for the treatment of sleep-related breathing disorders, including sleep apnea. The UIC License Agreement obligates the Company to comply with various commercialization and reporting requirements and to make various royalty payments, including potential one-time and annual royalty payments, as well as payments upon the achievement of certain development milestones.
In addition, the Company and the University of Wisconsin-Milwaukee Research Foundation, Inc., an affiliate of the University of Wisconsin-Milwaukee (“UWMRF”) executed the UWMRF Patent License Agreement effective August 1, 2020 pursuant to which RespireRx licensed the intellectual property identified therein, including with respect to GABAkines. In consideration for the licenses granted, the Company is required to pay to UWMRF patent filing and prosecution costs, annual license maintenance fees, one-time milestone payments, and annual royalties.
If we are unable to comply with the terms of these licenses, such as required payments thereunder, these licenses might be terminated and we would lose access to the licensed technologies or applications, which would have a material adverse effect on the Company’s ability to conduct research and development and operate.
We may not be able to successfully develop and commercialize our product candidates and technologies.
The development of our product candidates is subject to risks commonly experienced in the development of products based upon innovative technologies and the expense and difficulty of obtaining approvals from regulatory agencies. Drug discovery and development is time consuming, expensive and unpredictable. On average, only one out of many thousands of chemical compounds discovered by researchers proves to be both medically effective and safe enough to become an approved medicine.
All of our product candidates are in development spectrum that runs from preclinical to Phase 2 clinical trials, but we not have any currently active trials. Assuming these trials are initiated, which will require additional financing, we are planning for additional preclinical studies and Phase 1, Phase 2A, Phase 2B and Phase 3 clinical trials, we do not have any currently active trials. Accordingly, we will require significant additional funding for research, development and clinical testing of our product candidates, which may not be available on favorable terms or at all.
Additionally, our success, at least in part, is dependent upon the strength of our intellectual property, including, but not limited to licensed and owned patents, patent applications, continuations-in-part, provisional patent applications, know-how, trade secrets and other forms of intellectual property. The issuance of patents with relevant claims is subject to varying degrees of uncertainty. Our ability to defend our intellectual property or challenge third party intellectual property infringement claims is expensive, time consuming and uncertain. If our patent applications do not issue with relevant claims or if we cannot defend our patents, or, as appropriate, challenge interfering patents or actions of third parties, or otherwise maintain our intellectual property, our business and operations will be adversely affected.
| 11 |
The process from discovery to development to regulatory approval can take several years and drug candidates can fail at any stage of the process. Late-stage clinical trials often fail to replicate results achieved in earlier studies. We cannot be certain that we will be able to successfully complete any of our research and development activities. One of our product candidates is based, at least in part, on the development of one or more new formulations and the repurposing of an approved drug, the development of which is inherently risky while others of our product candidates have never been approved for marketing by any regulatory bodies and are subject to substantial research and development risks. Concerns about the safety and efficacy of our product candidates could limit our future success.
Even if we do complete our research and development activities, we may not be able to successfully market any of the product candidates or be able to obtain the necessary regulatory approvals or assure that healthcare providers and payors will accept our product candidates. We also face the risk that any or all of our product candidates will not work as intended or that they will be unsafe, or that, even if they do work and are safe, that our product candidates will be uneconomical to manufacture and market on a large scale. Due to the extended testing and regulatory review process required before we can obtain marketing clearance, we do not expect to be able to commercialize any therapeutic drug for several years, either directly or through our corporate partners or licensees.
We have announced a restructuring plan to facilitate the financing of our business initiatives. We may not achieve some or all of the expected benefits of our restructuring plan and the restructuring may adversely affect our business.
We plan to incorporate as newly formed subsidiaries, what are currently identified divisions of the Company, namely, ResolutionRx and EndeavourRx, with the goals, among others, of improving our ability to finance those platforms and attract potential strategic partners. There can be no assurance that these goals or any of our intended goals will be achieved, and the restructuring may adversely affect our business.
We have not voluntarily implemented various corporate governance measures, in the absence of which stockholders may have more limited protections against interested director transactions, conflicts of interests and similar matters.
We have not adopted any corporate governance measures since our securities are not yet listed on a national securities exchange and we are not required to do so. We have not adopted corporate governance measures such as separate audit or other independent committees of our Board as we presently have only one independent director. For example, in the absence of audit, nominating and compensation committees comprised of at least a majority of independent directors, decisions concerning matters such as compensation packages to our senior officers and recommendations for director nominees may be made by a majority of directors who have an interest in the outcome of the matters being decided. You should bear in mind our current lack of corporate governance measures in formulating investment decisions.
The novel coronavirus (COVID-19) pandemic may negatively impact our ability to successfully develop and commercialize our product candidates and technologies and may ultimately affect our business, financial condition and results of operations.
Although the COVID-19 pandemic seems to be diminishing in the United States, new variants may arise and the impact in many foreign countries is still severe. Vaccination rates in the United States have not achieved the desired levels believed to be necessary to diminish the chance of a resurgence. As described in more detail below, the global pandemic may adversely affect our business in many ways.
The COVID-19 virus and the related pandemic continues to evolve, has created significant uncertainty and economic disruption, and has led to record levels of unemployment nationally. Numerous state and local jurisdictions had previously imposed, and those and others in the future may impose, shelter-in-place orders, quarantines, shut-downs of non-essential businesses, and similar government orders and restrictions on their residents to control the spread of COVID-19.
The COVID-19 pandemic and government responses thereto have made it very difficult to recruit clinical trial subjects and patients and to conduct clinical trials in general. Although somewhat less than in the height of the pandemic prior to vaccine availability, we expect the life sciences industry and clinical trial activity to continue to face challenges arising from quarantines, site closures, travel limitations, interruptions to the supply chain for investigational products and other considerations if site personnel or trial subjects become infected with or are significantly at risk of contracting COVID-19. These challenges may lead to difficulties in meeting protocol-specified procedures. Further, in response to the public health emergency, the FDA issued guidance in March and July 2020 that was updated on January 27, 2021, emphasizing that safety of trial participants is critically important. Decisions to continue or discontinue individual patients or the trial are expected to be made by trial sponsors in consultation with clinical investors and Institutional Review Boards, which may lead to the implementation of additional protocols such as COVID-19 screening procedures, resulting in potential delays and additional costs. The risks, strategic and operational challenges and costs of conducting such trials as a result of the global pandemic have exacerbated an already challenging clinical trial process, which may negatively impact our ability to plan or conduct trials if we secure sufficient financing to enable us to pursue such activity.
In addition, we may be impacted by the downturn in the U.S. economy, which could have an adverse impact on our ability to raise capital and our business operations.
| 12 |
The extent to which COVID-19 ultimately impacts our business, financial condition and results of operations will depend on future developments, which are highly uncertain and unpredictable, including new information which may emerge concerning the severity and duration of the COVID-19 pandemic and the effectiveness of actions taken to contain the COVID-19 pandemic or treat its impact, among others. Additionally, the extent to which COVID-19 ultimately impacts our operations will depend on a number of factors, many of which will be outside of our control. The COVID-19 pandemic is evolving and new information emerges regularly, including for example, the FDA’s and other governmental regulatory bodies’ approval of various COVID-19 vaccinations products which are being widely distributed and administered in the United States and around the world; accordingly, the ultimate consequences of the COVID-19 pandemic cannot be predicted with certainty. In addition to the disruptions adversely impacting our business and financial results, they may also have the effect of heightening many of the other risks described in these risk factors, including risks relating to our ability to begin to generate revenue, to generate positive cash flow, our relationships with third parties, and many other factors. We will attempt to minimize these impacts, but there can be no assurance that we will be successful in doing so.
We may not be able to enter into the strategic alliances necessary to fully develop and commercialize our products and technologies, and we will be dependent on our strategic partners if we do.
We are seeking pharmaceutical companies and other strategic partners to participate with us in the development of major indications for the cannabinoids and neuromodulator compounds. These agreements would potentially provide us with additional funds or in-kind services in exchange for exclusive or non-exclusive license or other rights to the technologies and products that we are currently developing. Competition between biopharmaceutical companies for these types of arrangements is intense. We cannot give any assurance that our discussions with candidate companies will result in an agreement or agreements in a timely manner, or at all. Additionally, we cannot assure you that any resulting agreement will generate sufficient revenues to offset our operating expenses and longer-term funding requirements.
If our third-party manufacturers’ facilities do not follow established current good manufacturing guidelines and practices, our product development and commercialization efforts may be harmed.
There are a limited number of manufacturers that operate under the FDA’s and European Union’s good manufacturing practices regulations and are capable of manufacturing products like those we are developing. Third-party manufacturers may encounter difficulties in achieving quality control and quality assurance and may experience shortages of qualified personnel. A failure of third-party manufacturers to follow current good manufacturing practices or other regulatory requirements and to document their adherence to such practices may lead to significant delays in the availability of products for commercial use or clinical study, the termination of, or hold on, a clinical study, or may delay or prevent filing or approval of marketing applications for our products. In addition, we could be subject to sanctions, including fines, injunctions and civil penalties. Changing manufacturers may require additional clinical trials and the revalidation of the manufacturing process and procedures in accordance with FDA mandated current good manufacturing practices and would require FDA approval. This revalidation may be costly and time consuming. If we are unable to arrange for third-party manufacturing of our products, or to do so on commercially reasonable terms, we may not be able to complete development or marketing of our products.
Our ability to use our net operating loss carry forwards will be subject to limitations upon a change in ownership, which could reduce our ability to use those loss carry forwards following any change in Company ownership.
Generally, a change of more than 50% in the ownership of a Company’s stock, by value, over a three-year period constitutes an ownership change for U.S. federal income tax purposes. An ownership change may limit our ability to use our net operating loss carry forwards attributable to the period prior to such change. We have sold or otherwise issued shares of our common stock in various transactions sufficient to constitute an ownership change. As a result, if we earn net taxable income in the future, our ability to use our pre-change net operating loss carry forwards to offset U.S. federal taxable income will be subject to limitations, which would restrict our ability to reduce future tax liability. Future shifts in our ownership, including transactions in which we may engage, may cause additional ownership changes, which could have the effect of imposing additional limitations on our ability to use our pre-change net operating loss carry forwards.
Risks related to our industry
If we fail to secure adequate intellectual property protection, it could significantly harm our financial results and ability to compete.
Our success will depend, in part, on our ability to obtain and maintain patent protection for our products and processes in the United States and elsewhere. We have filed and intend to continue to file patent applications as we need them. However, additional patents that may issue from any of these applications may not be sufficiently broad to protect our technology. Also, any patents issued to us or licensed by us may be designed around or challenged by others, and if such design or challenge is effective, it may diminish our rights and negatively affect our financial results.
If we are unable to obtain and maintain sufficient protection of our proprietary rights in our products or processes prior to or after obtaining regulatory clearances, our competitors may be able to obtain regulatory clearance and market similar or competing products by demonstrating at a minimum the equivalency of their products to our products. If they are successful at demonstrating at least the equivalency between the products, our competitors would not have to conduct the same lengthy clinical tests that we have or will have conducted.
| 13 |
We also rely on trade secrets and confidential information that we protect by entering into confidentiality agreements with other parties. Those confidentiality agreements could be breached, and our remedies may be insufficient to protect the confidential information. Further, our competitors may independently learn our trade secrets or develop similar or superior technologies. To the extent that our consultants, key employees or others apply technological information independently developed by them or by others to our projects, disputes may arise regarding the proprietary rights to such information or developments. We cannot assure you that such disputes will be resolved in our favor.
We may be subject to potential product liability claims. One or more successful claims brought against us could materially adversely affect our business and financial condition.
The clinical testing, manufacturing and marketing of our products may expose us to product liability claims. We have never been subject to a product liability claim, and we require each patient in our clinical trials to sign an informed consent agreement that describes the risks related to the trials, but we cannot assure you that the coverage limits of our insurance policies will be adequate or that one or more successful claims brought against us would not have a material adverse effect on our business, financial condition and result of operations. Further, if one of our cannabinoid or AMPAkine compounds is approved by the FDA for marketing, we cannot assure you that adequate product liability insurance will be available, or if available, that it will be available at a reasonable cost. Any adverse outcome resulting from a product liability claim could have a material adverse effect on our business, financial condition and results of operations.
We face intense competition, and our competitors may develop products that are superior to those we are developing.
The pharmaceutical industry is characterized by intensive research efforts, rapidly advancing technologies, intense competition and a strong emphasis on proprietary therapeutics. Our competitors include many companies, research institutes and universities that are working in a number of pharmaceutical or biotechnology disciplines to develop therapeutic products similar to those we are currently investigating. Most of these competitors have substantially greater financial, technical, manufacturing, marketing, distribution or other resources than we do. In addition, many of our competitors have experience in performing human clinical trials of new or improved therapeutic products and obtaining approvals from the FDA and other regulatory agencies. We have no experience in conducting and managing later-stage clinical testing or in preparing applications necessary to obtain regulatory approvals. We expect that competition in this field will continue to intensify.
Our patents and patent applications do not cover the entire world, thus limiting the potential exclusive commercialization of our products to those countries in which we have intellectual property protection. We are aware of at least one company that may be developing a product or product similar to one of our prospective products for our proposed indication in countries where we do not have intellectual property protection. Such company or companies may choose to compete with us in countries where we do have intellectual property protection and cause us to expend resources defending our intellectual property. A liberal regulatory environment or unenforced or poorly enforced regulations may encourage competition from non-drug products such as medical cannabis or dietary supplements and similar products containing cannabis-derived molecules making claims that would be competitive with our proposed regulatory-approved claims. Since our target markets are very large, there is a great deal of economic incentive for others to enter and compete in those markets. We must compete with other companies with respect to their research and development efforts and for capital and other forms of funding. An inability to compete would have a material adverse impact on our business operations.
We may be unable to recruit and retain our senior management and other key technical personnel on whom we are dependent.
We are highly dependent upon senior management and key technical personnel and currently do not carry any insurance policies on such persons. In particular, we are highly dependent on Timothy L. Jones, our CEO and President, Arnold S. Lippa, Ph.D., our Chief Scientific Officer and Executive Chairman, and Jeff E. Margolis, our Senior Vice President, Chief Financial Officer, Treasurer and Secretary. Competition for qualified employees among pharmaceutical and biotechnology companies is intense. The loss of any of our senior management or other key employees, or our inability to attract, retain and motivate the additional or replacement highly skilled employees and consultants that our business requires, could substantially hurt our business prospects.
The regulatory approval process is expensive, time consuming, uncertain and may prevent us from obtaining required approvals for the commercialization of some of our products.
The FDA and other similar agencies in foreign countries have substantial requirements for therapeutic products. Such requirements often involve lengthy and detailed laboratory, clinical and post-clinical testing procedures and are expensive to complete. It often takes companies many years to satisfy these requirements, depending on the complexity and novelty of the product. The review process is also extensive, which may delay the approval process even more.
As of yet, we have not obtained any approvals to market our products. Further, we cannot assure you that the FDA or other regulatory agency will grant us approval for any of our products on a timely basis, if at all. Even if regulatory clearances are obtained, a marketed product is subject to continual review, and later discovery of previously unknown problems may result in restrictions on marketing or withdrawal of the product from the market.
| 14 |
Risks related to capital structure
Our stock price is volatile and our common stock could decline in value.
Our Common Stock is currently quoted for public trading on the OTCQB. The trading price of our Common Stock has been subject to wide fluctuations and may fluctuate in response to a number of factors, many of which will be beyond our control.
The market price of securities of life sciences companies in general has been very unpredictable. Broad market and industry factors may adversely affect the market price of our Common Stock, regardless of our operating performance. In the past, following periods of volatility in the market price of a company’s securities, securities class-action litigation has often been instituted. Such litigation, if instituted, could result in substantial costs for us and a diversion of management’s attention and resources.
The range of sales prices of our common stock, as adjusted for the reverse stock-split effected on January 5, 2021, for the fiscal years ended December 31, 2020 and 2019, as quoted on the OTC Markets, was $1.499 and $0.0200 and $8.5000 to $0.9800, respectively. The following factors, in addition to factors that affect that market generally, could significantly affect our business, and the market price of our common stock could decline:
| ● | competitors announcing technological innovations or new commercial products; | |
| ● | competitors’ publicity regarding actual or potential products under development; | |
| ● | regulatory developments in the United States and foreign countries; | |
| ● | legal developments regarding cannabinoids and cannabis products in the United States and foreign countries | |
| ● | developments concerning proprietary rights, including patent litigation; | |
| ● | public concern over the safety of therapeutic products; and | |
| ● | changes in healthcare reimbursement policies, healthcare regulations and standard of care requirements. |
Our common stock is thinly traded and you may be unable to sell some or all of your shares at the price you would like, or at all, and sales of large blocks of shares may depress the price of our common stock.
Our common stock has historically been sporadically or “thinly-traded,” meaning that the number of persons interested in purchasing shares of our common stock at prevailing prices at any given time may be relatively small or nonexistent. As a consequence, there may be periods of several days or more when trading activity in shares of our common stock is minimal or non-existent, as compared to a seasoned issuer that has a large and steady volume of trading activity that will generally support continuous sales without an adverse effect on share price. This could lead to wide fluctuations in our share price. You may be unable to sell your common stock at or above your purchase price, which may result in substantial losses to you. Also, as a consequence of this lack of liquidity, the trading of relatively small quantities of shares by our stockholders may disproportionately influence the price of shares of our common stock in either direction. The price of shares of our common stock could, for example, decline precipitously in the event a large number of shares of our common shares are sold on the market without commensurate demand, as compared to a seasoned issuer which could better absorb those sales without adverse impact on its share price.
There is a large number of shares of the Company’s common stock that may be issued or sold, and if such shares are issued or sold, the market price of our common stock may decline.
As of September 30, 2021, we had 90,396,596 shares of our common stock outstanding on a post-reverse stock split basis which occurred on January 5, 2021.
If all warrants and options outstanding as of September 30, 2021, were exercised prior to their respective expiration dates, up to 59,505,140 additional shares of our common stock could become freely tradable. The issuance of such shares would dilute the interests of the current stockholders and sales of substantial amounts of common stock in the public market could adversely affect the prevailing market price of our common stock and could also make it more difficult for us to raise funds through future offerings of common stock.
As of September 30, 2021, there were remaining outstanding convertible notes totaling $1,005,993 inclusive of accrued interest. Of that amount, $960,594 was convertible into 40,542,856 shares of common stock and $45,399 was convertible into an indeterminate number of shares of common stock as such notes may convert, at the option of each note holder, acting separately and independently of the other note holders, into the next exempt private securities offering of equity securities.
If we issue additional equity or equity-based securities, the number of shares of our common stock outstanding could increase substantially, which could adversely affect the prevailing market price of our common stock and could also make it more difficult for us to raise funds through future offerings of common stock.
| 15 |
Our charter document and other governing documents may prevent or delay an attempt by our stockholders to replace or remove management.
Certain provisions of our restated certificate of incorporation, as amended, could make it more difficult for a third party to acquire control of our business, even if such change in control would be beneficial to our stockholders. Our restated certificate of incorporation, as amended, allows the Board of Directors of the Company to issue, as of October 7, 2021, up to 5,000,000 shares of preferred stock, with characteristics to be determined by the board, without stockholder approval. The ability of our Board of Directors to issue additional preferred stock may have the effect of delaying or preventing an attempt by our stockholders to replace or remove existing directors and management. Section 203 of the Delaware General Corporation Law, from which we did not elect to opt out, provides that if a holder acquires 15% or more of our stock without prior approval of our Board of Directors, that holder will be subject to certain restrictions on its ability to acquire us within three years. These provisions may delay or deter a change in control of us, and could limit the price that investors might be willing to pay in the future for shares of our Common Stock.
Historically, warrants to purchase Common Stock have been issued as compensation for professional services, typically related to fund raising or have been issued in connection with the issuance of certain notes.
In addition, on several occasions, certain executive officers, members of the Board of Directors and certain vendors have offered to forgive accrued compensation and other amounts due to them, and the Board of Directors accepted such offers in exchange for either shares of Common Stock or options to purchase Common Stock. In particular, if executive officers offered and if the Board of Directors accepts such offer(s) in the future, a significant number of shares of Common Stock or one or more options to purchase a significant number of shares of Common Stock could be issued or granted. The ability of our Board of Directors to issue additional shares of Common Stock or options to purchase shares of Common Stock, or warrants to purchase shares of Common Stock, may have the effect of delaying or preventing an attempt by our stockholders to replace or remove existing directors and management.
If our common stock is determined to be a “penny stock,” a broker-dealer may find it more difficult to trade our common stock and an investor may find it more difficult to acquire or dispose of our common stock in the secondary market.
In addition, our common stock is subject to the so-called “penny stock” rules. The United States Securities and Exchange Commission (“SEC”) has adopted regulations that define a “penny stock” to be any equity security that has a market price per share of less than $5.00, subject to certain exceptions, such as any securities listed on a national securities exchange. For any transaction involving a “penny stock,” unless exempt, the rules impose additional sales practice requirements on broker-dealers, subject to certain exceptions. If our common stock is determined to be a “penny stock,” a broker-dealer may find it more difficult to trade our common stock and an investor may find it more difficult to acquire or dispose of our common stock on the secondary market.
We may issue additional shares of our Common Stock, and investment in our company is likely to be subject to substantial dilution.
Stockholders’ interests in the Company will be diluted and stockholders may suffer dilution in their net book value per share when we issue additional shares. Dilution is the difference between what investors pay for their stock and the net tangible book value per share immediately after the additional shares are purchased. We are authorized to issue up to 2,000,000,000 (2 billion) shares of Common Stock. Our financing activities in the past focused on convertible note financing that requires us to issue shares of Common Stock to satisfy principal, interest and any applicable penalties related to these convertible notes. When required under the terms and conditions of the convertible notes, we issue additional shares of Common Stock that have a dilutive effect on our stockholders. We anticipate that all or at least a substantial portion of our future funding, if any, will be in the form of equity financing from the sale of our Common Stock and so any investment in the Company will likely be diluted, with a resulting decline in the value of our Common Stock.
Additional financing may not be available on terms acceptable to us, and our ability to raise capital through equity financing may be limited by the number of authorized shares of our Common Stock. In order to raise significant additional amounts from equity financing, we will need to seek, and have sought, stockholder approval to amend our Certificate of Incorporation to increase the number of authorized shares of our Common Stock, and any such amendment would require the approval of the holders of a majority of the outstanding shares of our Common Stock. If we are unable to obtain needed financing on acceptable terms, we may not be able to implement our business plan, which could have a material adverse effect on our business, financial condition, results of operations and prospects.
Delaware law, our Certificate of Incorporation and our Bylaws provide for the indemnification of our officers and directors at our expense, and correspondingly limits their liability, which may result in a major cost to us and hurt the interests of our shareholders because corporate resources may be expended for the benefit of officers and/or directors.
Our Certificate of Incorporation and By-Laws of the Company, as amended (the “Bylaws”) include provisions that eliminate the personal liability of our directors for monetary damages for breach of fiduciary duty as a director, except for liability (i) for any breach of the director’s duty of loyalty to the Company or its stockholders, (ii) for acts or omissions not in good faith or which involve intentional misconduct or a knowing violation of law, (iii) under Section 174 of the Delaware General Corporation Law, or (iv) for any transaction from which the director derived an improper personal benefit. These provisions eliminate the personal liability of our directors and our shareholders for monetary damages arising out of any violation of a director of his fiduciary duty of due care, but do not affect a director’s liabilities under the federal securities laws or the recovery of damages by third parties.
Insofar as indemnification for liabilities arising under the Securities Act may be permitted to directors, officers or persons controlling the Company pursuant to provisions of the Delaware General Corporation Law, the Company has been informed that, in the opinion of the Securities and Exchange Commission, such indemnification is against public policy as expressed in that Act and is, therefore, unenforceable.
| 16 |
We do not intend to pay cash dividends on any investment in the shares of stock of our Company and any gain on an investment in our Company will need to come through an increase in our stock’s price, which may never happen.
We have never paid any cash dividends and currently do not intend to pay any cash dividends for the foreseeable future. To the extent that we require additional funding currently not provided for, our funding sources may prohibit the payment of a dividend. Because we do not currently intend to declare dividends, any gain on an investment in our Company will need to come through an increase in our Common Stock’s price. This may never happen, and investors may lose all of their investment in our Company.
FINRA sales practice requirements may also limit a stockholder’s ability to buy and sell our stock.
In addition to the “penny stock” rules described above, the Financial Industry Regulatory Authority (“FINRA”) has adopted rules that require that in recommending an investment to a customer, a broker-dealer must have reasonable grounds for believing that the investment is suitable for that customer. Prior to recommending speculative low-priced securities to their non-institutional customers, broker-dealers must make reasonable efforts to obtain information about the customer’s financial status, tax status, investment objectives and other information. Under interpretations of these rules, FINRA believes that there is a high probability that speculative low-priced securities will not be suitable for at least some customers. FINRA requirements make it more difficult for broker-dealers to recommend that their customers buy our Common Stock, which may limit your ability to buy and sell our stock and have an adverse effect on the market for our shares.
Costs and expenses of being a reporting company under the Exchange Act are substantial and may continue to impede us from ever achieving profitability.
We are subject to the reporting requirements of the Exchange Act and aspects of the Sarbanes-Oxley Act. We expect that the requirements of these rules and regulations will continue to comprise a substantial portion of our legal, accounting and financial compliance costs, and to make some activities more difficult, time-consuming and costly, placing significant strain on our personnel, systems and resources.
If we fail to remain current on our SEC reporting requirements, we could be removed from the OTCQB Venture Market, which would limit the ability of broker-dealers to sell our Common Stock and the ability of stockholders to sell their Common Stock in the secondary market.
Companies trading on the OTCQB Venture Market must be reporting issuers under Section 12 of the Exchange Act, must be current in their filings under the Exchange Act, and must meet continued listing requirements to maintain price quotation privileges on the OTCQB Venture Market. On December 10, 2020, our Common Stock was downlisted from the OTCQB Venture Market to the OTC Pink Sheets, because our Common Stock did not have a closing bid price of at least $0.01 per share once during a period of 30 consecutive trading days. On February 8, 2021, our Common Stock was uplisted to the OTCQB Venture Market, after our Common Stock underwent a ten-to-one (10:1) reverse stock split and after complying with the OTC Markets uplisting requirements. The OTCQB Venture Market is recognized by the SEC as an established public market.
In the future, if we fail to remain current on our reporting requirements, or otherwise do not meet listing requirements, we could be downlisted from the OTCQB Venture Market to the OTC Pink Sheets. The OTC Pink Sheets is the lowest and most speculative of the three over-the-counter marketplaces, and securities on the OTC Pink Sheets are more thinly and infrequently traded due to the more limited ability of broker-dealers and stockholders to buy or sell such securities. Accordingly, if we were forced to trade on the OTC Pink Sheets, the market for and liquidity of our Common Stock would be significantly diminished, and our ability to raise capital would be adversely impacted.
As a smaller reporting company and a non-accelerated filer, we are subject to scaled disclosure requirements that may make it more challenging for investors to analyze our results of operations and financial prospects and may cause investors to find our Common Stock less attractive.
As a smaller reporting company, we are subject to scaled disclosure requirements that may make it more challenging for investors to analyze our results of operations and financial prospects. For instance, as a “smaller reporting company,” which is generally defined as a company with less than $250 million of public float or a company with less than $100 million in annual revenues and either no public float or a public float of less than $700 million, we may elect to provide simplified executive compensation disclosures in our filings and take advantage of other decreased disclosure obligations in our filings with the SEC, including being required to provide only two years of audited financial statements in our annual reports. Consequently, it may be more challenging for investors to analyze our results of operations and financial prospects. Additionally, under current SEC rules, we are not an “accelerated filer” and so not required to include an auditor attestation of the effectiveness of our internal control over financial reporting in our annual reports on Form 10-K. We cannot predict if investors will find our Common Stock less attractive because we may rely on these reduced requirements. If some investors find our Common Stock less attractive as a result, there may be a less active trading market for our Common Stock and the price of shares of our Common Stock may be more volatile.
| 17 |
Assuming the sale of 375,000,000 shares of common stock at the offering price of $0.02 per share, we estimate that net proceeds will be approximately $6,760,000, after deducting estimated placement agent fees of $560,000 and estimated offering expenses of $180,000. We cannot assure you that this offering will be completed or that we will sell all of the 375,000,000 Shares.
We intend to use the net proceeds of this offering for general corporate purposes, which may include, but are not limited to, research and development expenditures, payment of existing accounts payable and accrued expenses obligations, supporting the execution of our strategy to separate our pharmaceutical cannabinoids and our neuromodulators programs into two new subsidiaries, as well as other general corporate and working capital purposes. Additionally, on August 31, 2021 and October 7, 2021, the Company issued two convertible notes with principal amounts of $115,000 each and an aggregate principal amount of $230,000 (the “Bridge Notes”). Interest on the Bridge Notes accrues at 10% per annum. The Bridge Notes mature on August 31, 2022 and October 7, 2022, respectively. The Bridge Notes may be prepaid, inclusive of guaranteed interest, subject to a 115% prepay premium until February 28, 2022 and April 7, 2022, respectively. The use of proceeds from the Bridge Notes was and is to pay certain expenses associated with this Offering, certain accounts payable and accrued expenses that relate to both research and development and general and administrative expenditures and working capital. We intend to use up to approximately $291,000 from the net proceeds of this Offering to prepay or repay a portion of the principal amount and accrued interest on the Bridge Notes over the next twelve (12) months, unless, in the alternative, a Bridge Note holder subscribes to the Offering and we permit such holder to apply outstanding principal and accrued interest on the Bridge Note to purchase Shares in the Offering, subject to the qualification of the Offering Statement of which this Offering Circular is a part and the Company’s receipt and acceptance of a Subscription Agreement from the Bridge Note holder. Management currently expects to deploy the net cash proceeds (assuming all Shares are sold for cash and no Shares are issued in consideration for cancellation of the Bridge Note or other liabilities) as set forth below (in order of priority); however, management reserves the right to use the proceeds in a manner other than described below.
| Percentage of Maximum Raised | ||||||||||||||||||||
| 100% | 80% | 60% | 40% | 20% | ||||||||||||||||
| Research and Development (as further specified below) | $ | 3,875,092 | $ | 3,175,092 | $ | 2,475,000 | $ | 1,700,000 | $ | 669,000 | ||||||||||
| Dronabinol research and development | 400,000 | 400,000 | 250,000 | 250,000 | 175,000 | |||||||||||||||
| AMPAkines research and development | 1,525,000 | 1,150,000 | 1,100,000 | 750,000 | 150,000 | |||||||||||||||
| GABAkines research and development | 650,000 | 325,000 | 325,000 | 300,000 | 200,000 | |||||||||||||||
| General research and development operations | 1,300,092 | 1,300,092 | 800,000 | 400,000 | 144,000 | |||||||||||||||
| Payment of accounts payable | 1,000,000 | 1,000,000 | 500,000 | 300,000 | 155,000 | |||||||||||||||
| Payment in cash of convertible promissory note inclusive of guaranteed interest and prepayment premium | 291,000 | 291,000 | 291,000 | 291,000 | 291,000 | |||||||||||||||
| General and administrative and working capital | 1,594,000 | 898,908 | 704,000 | 284,000 | 65,000 | |||||||||||||||
| Total Net Proceeds | $ | 6,760,000 | $ | 5,365,000 | $ | 3,970,000 | $ | 2,575,000 | $ | 1,180,000 | ||||||||||
The Company cannot predict with certainty all of the particular uses for the proceeds from this Offering or the amounts that it will actually spend on the uses set forth above. The approximate amounts set forth above to describe the intended uses of proceeds are estimates subject to a variety of assumptions as well as trial design. The amounts and timing of the Company’s actual research and development expenditures will depend upon numerous factors, including the progress of these projects and the amount of funding actually raised. Accordingly, the Company’s management will have flexibility in applying the net proceeds of this Offering.
| 18 |
If you purchase shares in this Offering, your ownership interest in our Common Stock will be diluted immediately, to the extent of the difference between the price to the public charged for each share of Common Stock in the Unit in this Offering and the negative net tangible book value per share of our Common Stock after this Offering. In addition, you may be further diluted by conversions of convertible notes into Common Stock, forgiveness or exchange of accounts payable or accrued expenses for Common Stock, exercise of options and warrants and by future offerings of RespireRx’s Common Stock or other equity-linked securities or instruments that may convert or exercise into Common Stock.
As of September 30, 2021, our total negative net tangible book value was $9,424,888 and, based on 90,396,596 shares of Common Stock outstanding as of September 30, 2021, our negative net tangible book value per share was $0.1043. Therefore, on a pro forma basis, if you were charged $0.02 per share in the Offering and if the maximum number of shares offered were sold, you would experience dilution of $0.0257 per share.
The following tables illustrate the per share dilution to new investors discussed above, assuming the sale of, respectively, 100%, 80%, 60%, 40% and 20% of the shares offered for sale in this Offering (after our estimated placement agent fees of 7% of the assumed gross proceeds and estimated offering expenses of $180,000) and utilizing the negative net tangible book value and the number of outstanding shares of Common Stock as of September 30, 2021:
| Percentage of Maximum Offering issued | 100% | 80% | 60% | 40% | 20% | |||||||||||||||
| Funding Level | $ | 6,760,000 | $ | 5,365,000 | $ | 3,970,000 | $ | 2,575,000 | $ | 1,180,000 | ||||||||||
| Offering Price per share calculated at maximum of range | $ | 0.0200 | $ | 0.0200 | $ | 0.0200 | $ | 0.0200 | $ | 0.0200 | ||||||||||
| Net tangible book value per share of Common Stock before the Offering | $ | (0.1043 | ) | $ | (0. 1043 | ) | $ | (0. 1043 | ) | $ | (0. 1043 | ) | $ | (0. 1043 | ) | |||||
| Increase per common share attributable to investors in this Offering | $ | 0.0985 | $ | 0.0939 | $ | 0.0870 | $ | 0.0758 | 0.0544 | |||||||||||
| Pro forma net tangible book value per share of Common Stock after the Offering | $ | (0.0057 | ) | $ | (0.0104 | ) | $ | (0.0173 | ) | $ | (0.0285 | ) | $ | (0.0498 | ) | |||||
| Pro forma net tangible book value after the offering | $ | (2,664,888 | ) | $ | (4,059,888 | ) | $ | (5,454,888 | ) | $ | (6,849,888 | ) | $ | (8,244,888 | ) | |||||
| Dilution to investors | $ | 0.0257 | $ | 0.0304 | $ | 0.0373 | $ | 0.0485 | $ | 0.0698 | ||||||||||
| Dilution as a percent of Offering Price | 128.36 | % | 152.00 | % | 186.48 | % | 242.47 | % | 349.25 | % |
| 19 |
The following tables set forth, assuming the sale of, respectively, 100%, 80%, 60%, 40% and 20% of the Shares offered for sale in this Offering, the total number of shares previously sold to existing stockholders, the total consideration paid for the foregoing and the respective percentages applicable to such purchased shares and consideration paid, based on $0.02 per share paid by investors in this Offering utilizing the total consideration paid by existing shareholders as of September 30, 2021.
| Shares | Purchased | Total Cash | Consideration | |||||||||||||
| Number | Percentage | Amount | Percentage | |||||||||||||
| Assuming 100% of shares sold | ||||||||||||||||
| Existing stockholders | 90,396,596 | 19.42 | % | $ | 163,634,175 | 95.62 | % | |||||||||
| New Investors | 375,000,000 | 80.58 | % | 7,500,000 | 4.38 | % | ||||||||||
| Total | 465,396,596 | 100.00 | % | $ | 171,134,175 | 100.00 | % | |||||||||
| Shares | Purchased | Total Cash | Consideration | |||||||||||||
| Number | Percentage | Amount | Percentage | |||||||||||||
| Assuming 80% of shares sold | ||||||||||||||||
| Existing stockholders | 90,396,596 | 23.16 | % | $ | 163,634,175 | 96.46 | % | |||||||||
| New Investors | 300,000,000 | 76.84 | % | 6,000,000 | 3.54 | % | ||||||||||
| Total | 390,396,596 | 100.00 | % | $ | 169,634,175 | 100.00 | % | |||||||||
| Shares | Purchased | Total Cash | Consideration | |||||||||||||
| Number | Percentage | Amount | Percentage | |||||||||||||
| Assuming 60% of shares sold | ||||||||||||||||
| Existing stockholders | 90,396,596 | 28.66 | % | $ | 163,634,175 | 97.32 | % | |||||||||
| New Investors | 225,000,000 | 71.34 | % | 4,500,000 | 2.68 | % | ||||||||||
| Total | 315,396,596 | 100.00 | % | $ | 168,134,175 | 100.00 | % | |||||||||
| Shares | Purchased | Total Cash | Consideration | |||||||||||||
| Number | Percentage | Amount | Percentage | |||||||||||||
| Assuming 40% of shares sold | ||||||||||||||||
| Existing stockholders | 90,396,596 | 37.60 | % | $ | 163,634,175 | 98.20 | % | |||||||||
| New Investors | 150,000,000 | 62.40 | % | 3,000,000 | 1.80 | % | ||||||||||
| Total | 240,396,596 | 100.00 | % | $ | 166,634,175 | 100.00 | % | |||||||||
| Shares | Purchased | Total Cash | Consideration | |||||||||||||
| Number | Percentage | Amount | Percentage | |||||||||||||
| Assuming 20% of shares sold | ||||||||||||||||
| Existing stockholders | 90,396,596 | 54.65 | % | $ | 163,634,175 | 99.09 | % | |||||||||
| New Investors | 75,000,000 | 45.35 | % | 1,500,000 | 0.91 | % | ||||||||||
| Total | 165,396,596 | 100.00 | % | $ | 165,134,175 | 100.00 | % | |||||||||
The foregoing discussion and tables are based on 90,396,596 shares of common stock outstanding and the historical net tangible book value calculation as of September 30, 2021, which gives effect to the pro forma transactions described above, and excludes as of such date (unless otherwise specified):
| ● | 7,111,924 shares issuable upon the exercise of outstanding stock options as of September 30, 2021, | |
| ● | 15,757,542 shares reserved and available for future issuances under our equity plans as of September 30, 2021, | |
| ● | 59,505,140 shares issuable upon the exercise of stock purchase warrants outstanding*, | |
| ● | 40,542,857 shares issuable upon conversion of convertible promissory notes outstanding*, and | |
| ● | 649 shares issuable as “Pier Contingent Shares.” |
* Does not include or account for (a) shares issuable upon conversion of additional interest accrued after September 30, 2021, (b) 5,750,000 shares issuable upon conversion of a convertible promissory note issued on October 7, 2021, or 5,750,000 shares issuable upon exercise of warrants issued on the same date in connection with such convertible promissory note, (c) 4,000,000 shares issued on November 8, 2021 upon partial conversion of $80,000 of a $112,000 convertible note, which reduced the principal amount of such note to $32,000 and reduced the number of shares into which such note is convertible by 4,000,000 shares, (d) the partial cashless exercise on November 8, 2021, of a warrant which reduced the number of warrants by 1,534,042 and resulted in the issuance of 511,347 shares of common stock or (e) the cashless exercise on November 22, 2021, of a warrant which reduced the number of warrants by 4,300,000 and resulted in the issuance of 1,433,333 shares of common stock.
(3) Excludes the Shares sold for cash or which may be issued as repayment of accounts payable, accrued expenses, principal on promissory notes, convertible or otherwise, inclusive or exclusive of interest, or other liabilities, all or any of which might occur without notice to subscribers, and would only occur pursuant to Subscription Agreements accepted by the Company. Only Shares offered for cash will be issued and sold at the first closing. Also excludes the shares of common stock issuable upon the exercise of warrants issued to the Placement Agent. See “Plan of Distribution.”
To the extent that stock options or warrants are exercised or convertible promissory notes are converted, new stock options are issued under our equity plan, or we issue additional shares of common stock in the future, there will be further dilution to investors participating in this Offering. In addition, we may choose to raise additional capital because of market conditions or strategic considerations, even if we believe that we have sufficient funds for our current or future operating plans. If we raise additional capital through the sale of equity or convertible debt securities, the issuance of these securities could result in further dilution to our stockholders.
| 20 |
General
We are offering up to 375,000,000,000 Shares on a “best efforts” basis. There is no minimum investment amount.
In addition this Offering Circular, we may use our existing website, www.respirerx.com, blogs, and other social media to provide notification of the Offering. Persons who desire information will be directed to a landing page operated by us where a copy of this Offering Circular may be downloaded. This Offering Circular and any supplemental disclosure or disclosures will be furnished to prospective investors via download 24 hours per day, 7 days per week on our website. This Offering Circular and any supplemental disclosure or disclosures are also available at www.SEC.gov. Prospective investors may also request copies of this Offering Circular and any supplemental disclosure or disclosures directly from us or from our Placement Agent, as defined the next sentence. Primary Capital LLC (“Primary Capital”), a broker-dealer registered with the SEC and a member of FINRA and Securities Investor Protection Corporation, is our Placement Agent (“Placement Agent”) for this Offering.
In order to subscribe to purchase the Shares, a prospective investor must complete a Subscription Agreement. Upon submission to RespireRx or the Placement Agent, RespireRx and/or the Placement Agent will review the submitted Subscription Agreement and the Company may accept or reject such Subscription Agreement in its sole discretion. If accepted, the subscribing investor will be provided payment instructions to send payment by wire transfer or ACH. The investor qualification questionnaire section of the Subscription Agreement requires investors to answer certain questions to determine if they are accredited investors as that term is defined in Rule 501 under the Securities Act and are eligible to invest in the Offering in the amount subscribed or are otherwise eligible to invest in the Offering.
There will be no escrow and investor funds will not be held in an escrow account. All subscribers will be instructed by us or our agents to transfer funds by wire or ACH transfer directly to one of our bank accounts. We may terminate the offering at any time for any reason at our sole discretion. Investors should understand that if they remit funds and their Subscription Agreement is not accepted, their funds will be returned and they will not be deemed an investor in the Company and their subscription will be deemed null and void ab initio.
We may close on investments on a “rolling” basis (so not all investors will receive their Shares on the same date). The Shares will be sold for cash or may be issued as repayment of accounts payable, accrued expenses, principal on promissory notes, convertible or otherwise, inclusive or exclusive of interest, or other liabilities, all or any of which might occur without notice to subscribers, and would only occur pursuant to Subscription Agreements accepted by the Company. There is a “Closing” each time we accept funds or the application of non-cash consideration in exchange for Shares. Only Shares offered for cash will be issued and sold at the first closing. Upon a Closing for funds tendered by investors, the funds will be immediately available to us for our use. The Offering will terminate at the earlier of: (a) the sale of all 375,000,000,000 Shares offered hereby, (b) October 12, 2023, or (c) when the Company’s board of directors elects to terminate the Offering.
Placement Agent and Placement Agent Compensation
We have engaged Primary Capital, LLC as our exclusive placement agent with respect to this Offering (“Primary Capital” or the “Placement Agent”) pursuant to an engagement agreement dated August 6, 2021, as amended on September 29, 2021 and October 12, 2021 (the “Engagement Agreement”).
Pursuant to the Engagement Agreement, Placement Agent has agreed to use its best efforts to procure potential purchasers for the shares of our Common Stock being offered as well as render certain other investment banking services set forth therein. This Offering is being undertaken on a best efforts basis only. Neither the Placement Agent nor any future selling group member that may be engaged is required to take or pay for any specific number or dollar amount of our common stock. The Placement Agent will have the right, subject to our approval, to engage such other FINRA member firms as it determines to assist in this offering.
The Placement Agent will offer the Shares only to institutional “accredited investors” (as defined and described in Rule 501(a)(1), (2), (3), and (7) of Regulation D of the Securities Act) who have prior experience in investing in penny stocks or have previously invested in the Company. We are also required to pay the Placement Agent a placement fee. At the Closing of each and any sale in the Offering, placement fees are calculated as follows:
| ● | For institutional accredited investors originated by the Placement Agent, we are required to pay the Placement Agent a cash fee equal to 7% of the gross proceeds from the Offering invested by such institutional investors, and issue the Placement Agent a warrant (a “Placement Agent Warrant”) exercisable for 7% of the Shares sold to such investors; and |
| ● | For institutional accredited investors referred by us or others to the Placement Agent, we are required to pay the Placement Agent a cash fee equal to 4% of the gross proceeds from the Offering invested by institutional investors, and to issue the Placement Agent a Placement Agent Warrant exercisable for 4% of the number of Shares sold to such persons. |
The following table shows the per Share and total cash commissions to be paid to the Placement Agent, assuming 375,000,000 shares of Common Stock are sold at the maximum price of $0.02 per share and all investors were originated by the Placement Agent:
| Commissions paid by the Company | ||||
| Per Share | $ | 0.0014 | ||
| Total | $ | 525,000 | ||
For non-institutional accredited investors originated by us through introductions made by our current shareholders, there will be no cash fee or warrants due to the Placement Agent.
In all cases, the Placement Agent Warrants will have an exercise price equal to 100% of the price at which the Shares are being sold in this offering. The Placement Agent Warrants will include, among other things, certain anti-dilution provisions providing that if the Company subdivides (by any stock split, reverse stock split, stock dividend, recapitalization or otherwise) one or more classes of its outstanding shares of Common Stock into a greater or lesser number of shares, the number of shares of Common Stock issuable upon the exercise of the Placement Agent Warrants and the exercise price will be adjusted proportionately. The Placement Agent Warrants will also include standard “cashless” exercise provisions.
The Placement Agent Warrants to be received by the Placement Agent and related persons in connection with this offering: (i) fully comply with lock-up restrictions pursuant to FINRA Rule 5110(e)(1); and (ii) fully comply with transfer restrictions pursuant to FINRA Rule 5110(e)(2).
Under FINRA Rule 5110(e)(1), the Placement Agent Warrants and any shares issued upon exercise of the Placement Agent Warrants shall not be sold, transferred, assigned, pledged or hypothecated, or be the subject of any hedging, short sale, derivative, put or call transaction that would result in the effective economic disposition of the securities for a period of 180 days beginning on the date of commencement of sales of this offering (the “Initial Exercise Date”), except as provided for in FINRA Rule 5110(e)(2) including:
| ● | by operation of law or by reason of reorganization of the Company; |
| ● | to any FINRA member firm participating in the offering and the officers or partners thereof, if all securities so transferred remain subject to the lock-up restriction set forth below for the remainder of the time period; |
| ● | if the aggregate amount of securities of the Company held by the holder of the underwriter purchase options or related persons do not exceed 1% of the securities being offered; |
| ● | that is beneficially owned on a pro rata basis by all equity owners of an investment fund; provided, that no participating member manages or otherwise directs investments by the fund, and participating members in the aggregate do not own more than 10% of the equity in the fund; or |
| ● | the exercise or conversion of any security, if all securities received remain subject to the lock-up restriction set forth above for the remainder of the time period. |
The Placement Agent Warrants will expire on the third anniversary of the Initial Exercise Date. The Placement Agent Warrants will not have any demand or piggyback registration rights.
Notwithstanding the foregoing, the Placement Agents Warrants will be in compliance with FINRA Rule 5110(g)(8), and in no event shall:
| (A) | be exercisable for more than five years from the commencement of sales of the offering; | |
| (B) | have more than one demand registration right at the issuer’s expense; have a demand registration right with a duration of more than five years from the commencement of sales of the public offering; | |
| (C) | have a piggyback registration right with a duration of more than seven years from the commencement of sales of the public offering; | |
| (D) | have anti-dilution terms that allow the participating members to receive more shares or to exercise at a lower price than originally agreed upon at the time of the public offering, when the public shareholders have not been proportionally affected by a stock split, stock dividend, or other similar event; or | |
| (E) | have anti-dilution terms that allow the participating members to receive or accrue cash dividends prior to the exercise or conversion of the security. |
In connection with our Engagement Agreement, we have previously paid the Placement Agent a non-accountable and non-refundable due diligence fee of $7,500 (USD).
We have also agreed to reimburse the Placement Agent for its reasonable legal expenses incurred in the performance of its services in an amount up to $25,000. We have agreed to reimburse the Placement Agent and its co-agents for other reasonable documented expenses incurred in the performance of its services, such expenses not to exceed $35,000, inclusive of the $25,000 for legal expenses incurred by the Placement Agent.
The term of the Engagement Agreement is 12 months (the “Engagement Period”). The Company or the Placement Agent can terminate the Engagement Agreement for any reason upon thirty (30) days written notice after the six (6) month anniversary of the Engagement Agreement, or February 6, 2021.
If during the 12-month period after the expiration or termination of the Engagement Agreement, the Company consummates any offering, licensing agreement or partnership, sale or acquisition with any party introduced to the Company by the Placement Agent during the Engagement Period, then the Company shall pay the Placement Agent the full consideration to which the Placement Agent would have been entitled to hereunder had the Engagement Agreement not expired or been terminated.
| 21 |
None.
Overview
The Company was incorporated in Delaware in 1987 as Cortex Pharmaceuticals, Inc. and changed its name to RespireRx Pharmaceuticals Inc. in 2015.
The mission of the Company is to develop innovative and revolutionary treatments to combat disorders caused by disruption of neuronal signaling. We are developing treatment options that address obstructive sleep apnea (“OSA”), attention deficit hyperactivity disorder (“ADHD”), epilepsy, chronic pain, including inflammatory and neuropathic pain, and recovery from spinal cord injury (“SCI”), which are conditions that affect millions of people but for which there are limited or poor treatment options. We are also considering developing treatment options for other conditions based on results of preclinical and clinical studies to date.
RespireRx is developing a pipeline of new drug products supported by our broad patent portfolios across two distinct drug platforms:
| (i) | our pharmaceutical cannabinoids platform (which we refer to as ResolutionRx) is developing compounds that target the body’s endocannabinoid system, and in particular, the re-purposing of dronabinol, an endocannabinoid CB1 and CB2 receptor agonist, for the treatment of OSA. Dronabinol is already approved by the FDA for other indications. | |
| (ii) | our neuromodulators platform (which we refer to as EndeavourRx) is made up of two programs: (a) our AMPAkines program, which is developing proprietary compounds that are positive allosteric modulators (“PAMs”) of AMPA-type glutamate receptors to promote neuronal function and (b) our GABAkines program, which is developing proprietary compounds that are PAMs of GABAA receptors, and which was recently established pursuant to our entry with the University of Wisconsin-Milwaukee Research Foundation, Inc., an affiliate of the University of Wisconsin-Milwaukee (“UWMRF”), into a patent license agreement (the “UWMRF Patent License Agreement”). |
In order to facilitate our business activities and product development, we have organized our drug platforms into two separate business units. The business unit focused on pharmaceutical cannabinoids is referred to as ResolutionRx and the business unit focused on neuromodulators is referred to as EndeavourRx. It is anticipated that the Company will use, at least initially, its management personnel to provide management, operational and oversight services to these two business units.
Management intends to organize our ResolutionRx and EndeavourRx business units into two subsidiaries: (i) a ResolutionRx subsidiary, into which we would contribute our pharmaceutical cannabinoid platform and its related tangible and intangible assets and certain of its liabilities and (ii) an EndeavourRx subsidiary, into which we would contribute our neuromodulator platform, including both the AMPAkine and GABAkine programs and their related tangible and intangible assets and certain of their liabilities.
Management believes that there are advantages to separating these platforms formally into newly formed subsidiaries, including but not limited to optimizing their asset values through separate financing channels and making them more attractive for capital raising as well as for strategic transactions.
The Company is also engaged in business development efforts (licensing/sub-licensing, joint venture and other commercial structures) with a view to securing strategic partnerships that represent strategic and operational infrastructure additions, as well as cash and in-kind funding opportunities. These efforts have focused on, but have not been limited to, transacting with brand and generic pharmaceutical and biopharmaceutical companies as well as companies with potentially useful formulation or manufacturing capabilities, significant subject matter expertise and financial resources. No assurance can be given that any transaction will come to fruition and that if it does, that the terms will be favorable to the Company.
| 22 |
Neurotransmission
RespireRx is developing drugs to modify neurotransmission and create advanced treatments for disorders with high unmet needs. Neurotransmission is the basic process in the brain by which specialized nerve cells called neurons communicate information with each other.
 |
As illustrated in this figure, during neurotransmission, neurons release chemicals called neurotransmitters which attach to receptors, very specific protein structures residing on adjacent neurons. This enables neurons to communicate with one another by either increasing or decreasing the excitability of the neuron receiving the communication. For example, glutamate is the primary excitatory neurotransmitter in the brain, while gamma-amino-butyric acid (“GABA”) is the primary inhibitory neurotransmitter. Neurons also contain receptors for anandamide (AEA) and 2-arachidonoylglycerol (2-AG), the brain’s own natural cannabinoid (endocannabinoid) neurotransmitters. |
ResolutionRx – Pharmaceutical Cannabinoids
Background
The term cannabinoid refers to pharmacologically active substances originally found within the cannabis plant that led to the discovery of the body’s own cannabinoids, termed endocannabinoids. Endocannabinoids are endogenous neurotransmitters located throughout the brain and peripheral nervous system that are used by certain nerve cells to convey information from cell to cell. The two major endocannabinoids that have been identified are anandamide (AEA) and 2-arachidonoylglycerol (2-AG), which are secreted and act upon CB1 and CB2 endocannabinoid receptors, thereby influencing a variety of physiological functions, including respiration, appetite, convulsions and potentially others.
Due to the liberalization of state laws regulating the use and sales of cannabis over the last 5 years, a major industry has grown around its commercialization. However, while cannabis use has been legalized in certain states, it still is not legal under federal statutes and regulations. The medical use of any pharmacological agent must be approved by the U.S Food and Drug Administration (“FDA”) and, to date, the FDA has not recognized or approved the cannabis plant as medicine nor is it federally legal to sell products that contain cannabinoids as drugs or dietary supplements without its approval.
Worldwide clinical research efforts have established the cannabinoid class of compounds as bona fide pharmaceutical products, or “pharmaceutical cannabinoids,” which are being developed and commercialized according to FDA regulatory and industry guidelines. Scientific research and commercial development to date has focused primarily on two major cannabinoids, dronabinol and cannabidiol (“CBD”). This research and development effort began in 1985 when dronabinol, a directly acting agonist on CB1 and CB2 receptors, was approved by the FDA as Marinol® for the treatment of AIDS-related anorexia and later for the treatment of chemotherapy-induced nausea and vomiting. Marinol®, as well as generic dronabinol, is available in 2.5 mg, 5 mg, and 10 mg capsules, with a maximum labelled dosage of 20 mg/day for the AIDS indication, or 15 mg/m2 per dose for chemotherapy-induced nausea and vomiting.
This breakthrough subsequently led to the 2018 FDA approval of Epidiolex®, a proprietary oral solution of CBD sold by GW Pharmaceuticals plc (“GW Pharma”) for the treatment of certain rare, treatment-resistant forms of epilepsy. Nabiximol®, an oromucosal spray containing Δ9-THC and CBD, was approved under the tradename Sativex® by applicable regulatory authorities in 29 countries outside the United States and is marketed and distributed by GW Pharmaceuticals plc (“GW”) (On May 5, 2021, GW and Jazz Pharmaceuticals plc (“Jazz”) announced the completion of Jazz’s acquisition of GW).
The commercialization of these pharmaceutical cannabinoids has opened the door to an expanding market sector. As part of our effort to capitalize upon this opportunity, the Company has implemented an internal restructuring plan by forming ResolutionRx as a business unit focused on the pharmaceutical cannabinoid market. ResolutionRx’s initial primary focus has been and will continue to be the re-purposing of dronabinol using new proprietary formulations and therapeutic indications. Because dronabinol already is an approved drug, we intend to use publicly available information, particularly safety data, in support of a 505(b)(2) New Drug Application (“NDA”), a potentially more rapid route to FDA approval than a standard 505(b)(1) NDA.
Obstructive Sleep Apnea (OSA)
The Company is developing dronabinol for the treatment of OSA, a sleep-related breathing disorder that afflicts an estimated 29 million people in the United States according to the American Academy of Sleep Medicine (“AASM”), and an additional 26 million in Germany and 8 million in the United Kingdom, as presented at the European Respiratory Society’s annual Congress in Paris, France in September 2018. OSA involves a decrease or complete halt in airflow despite an ongoing effort to breathe during sleep. When the muscles relax during sleep, soft tissue in the back of the throat collapses and obstructs the upper airway. OSA remains significantly under-recognized, as only 20% of cases in the United States according to the AASM and 20% of cases globally have been properly diagnosed. About 24 percent of adult men and 9 percent of adult women are believed to have the breathing symptoms of OSA with or without daytime sleepiness. OSA significantly impacts the lives of sufferers who do not get enough sleep; their quality of sleep is deteriorated such that daily function is compromised and limited. OSA is associated with decreased quality of life, significant functional impairment, and increased risk of road traffic accidents, especially in professions like road and rail transportation and shipping.
| 23 |
Research has established links between OSA and several important co-morbidities, including hypertension, type II diabetes, obesity, stroke, congestive heart failure, coronary artery disease, cardiac arrhythmias, and even early mortality. The consequences of undiagnosed and untreated OSA are medically serious and economically costly. According to the AASM, the estimated economic burden of OSA in the United States is approximately $162 billion annually. All current treatment options have serious drawbacks. We believe that a new drug therapy that is effective in reducing the medical and economic burden of OSA would have major benefits for the treatment of this costly disease indication.
Continuous Positive Airway Pressure (“CPAP”) is the most common treatment for OSA. CPAP devices work by blowing pressurized air into the nose (or mouth and nose), which keeps the pharyngeal airway open. Patients must use the device whenever they sleep. Reduction of the apnea-hypopnea index (“AHI”) is the standard objective measure of therapeutic response in OSA. Apnea is the cessation of breathing for 10 seconds or more and hypopnea is a reduction in breathing. AHI is the sum of apnea and hypopnea events per hour. In the sleep laboratory, CPAP is highly effective at reducing AHI. However, the device is cumbersome and difficult for many patients to tolerate. Most studies describe that 25-50% of patients refuse to initiate or completely discontinue CPAP use within the first several months and that most patients who continue to use the device do so only intermittently.
Oral devices may be an option for patients who cannot tolerate CPAP. Several dental devices are available. The cost of these devices tends to be high and side effects associated with them include night-time pain, dry lips, tooth discomfort, and excessive salivation.
Patients with clinically significant OSA who cannot be treated adequately with CPAP or oral devices may elect to undergo surgery, the most common form of which involves the removal of excess tissue in the throat to make the airway wider. Patients who undergo surgery for the treatment of OSA risk complications. Surgery is often unsuccessful, and at present, no method exists to reliably predict therapeutic outcome from surgery.
In 2014, another surgical option first became available based on upper airway stimulation. This was later followed by a second-generation medical device cleared by the FDA in 2017. It is a combination of an implantable nerve stimulator and an external remote controlled by the patient. The implanted device stimulates the hypoglossal nerve, which controls the tongue, with every attempted breath, regardless of whether such stimulation is needed for that breath. The device is turned on at night and off in the morning by the patient with the remote.
The Company’s Research Efforts Regarding the Treatment of OSA with Cannabinoids
The Company conducted a 21-day, randomized, double-blind, placebo-controlled, dose escalation Phase 2A clinical study in 22 patients with OSA, in which FDA approved and commercially available dronabinol produced a statistically significant reduction in AHI, the primary therapeutic end-point, and was observed to be safe and well tolerated, with the frequency of side effects no different from placebo. This clinical trial provided data supporting the submission of patent applications claiming unique dosage strengths, blood levels and controlled release formulations optimized for use in the treatment of OSA. If approved, these pending patents would extend market exclusivity until January 2042.
With approximately $5 million in funding from the National Heart, Lung and Blood Institute of the National Institutes of Health (“NIH”), Dr. David Carley of the University of Illinois at Chicago (“UIC”), along with his colleagues at UIC and Northwestern University, completed a Phase 2B multi-center, double-blind, placebo-controlled clinical trial of FDA approved and commercially available dronabinol in patients with OSA. This study, named “Pharmacotherapy of Apnea with Cannabimimetic Enhancement” (“PACE”) replicated our earlier Phase 2A study. The authors published in January 2018 in the journal SLEEP and reported that, in a dose-dependent fashion, treatment with 2.5 mg and 10 mg of dronabinol once per day at night significantly reduced, compared to placebo, AHI during sleep in 56 evaluable patients with moderate to severe OSA who completed the study. Additionally, treatment with 10 mg of dronabinol significantly improved daytime sleepiness as measured by the Epworth Sleepiness Scale and achieved the greatest overall patient satisfaction. As in our previous Phase 2A study, dronabinol was observed to be safe and well tolerated, with the frequency of side effects no different from placebo. The Company did not manage this clinical trial, which was funded entirely by the National Heart, Lung and Blood Institute of NIH.
The Opportunity to Improve Dronabinol Formulations
Dronabinol is currently marketed as a soft gelatin capsule that suffers from several major deficiencies.
First, dronabinol is not water soluble and exhibits poor and erratic absorption. The market-dominant commercial gel cap formulation of dronabinol is currently formulated as a sesame oil-based liquid within a soft gelatin capsule. The absorption of dronabinol after oral administration is poor and highly variable with some patients achieving very high levels and others achieving very low levels. This erratic absorption may be responsible for the variable therapeutic responses observed in dronabinol clinical trials. Syndros®, on the other hand, is formulated as a dronabinol solution in dehydrated alcohol, polyethylene glycol and other materials and exhibits its own challenges and deficiencies, including but not limited to it being classified as a Schedule II drug by the U.S. Drug Enforcement Administration (the “DEA”) as compared to the capsule formulation that is classified as a Schedule III drug.
| 24 |
Second, dronabinol is rapidly and extensively (approximately 80%) metabolized upon first pass through the liver, resulting in low blood levels. Additionally, dronabinol has a relatively short half-life (approximately 3 – 4 hours) and, in its present formulation, is not optimally suited for therapeutic indications requiring blood levels to be sustained for 6 hours or longer.
Third, in order to achieve sustained, therapeutic blood levels, we have found it necessary to use higher doses of dronabinol in our OSA clinical trials. For example, over an 8-hour period, the 2.5 mg and 10 mg doses produced therapeutically equivalent effects during the first 4 hours, but only the 10 mg dose produced therapeutic effects during the second 4 hours. Unfortunately, the 10 mg dose produces a higher occurrence of side effects than the 2.5 mg dose (as described in the Marinol® package insert). We are currently developing new formulations that would achieve the blood levels produced by the lower doses for a sustained time period, resulting in the desired therapeutic effect(s) while minimizing undesirable side effects.
The Company’s Cannabinoid Intellectual Property Rights
In order to expand RespireRx’s respiratory disorders program and develop cannabinoids for the treatment of OSA, RespireRx acquired 100% of the issued and outstanding equity securities of Pier Pharmaceuticals, Inc. (“Pier”) effective August 10, 2012 pursuant to an Agreement and Plan of Merger. Pier was a clinical stage pharmaceutical company developing a pharmacologic treatment for OSA and had been engaged in research and clinical development activities.
On June 27, 2014, RespireRx entered into an exclusive license agreement (the “2014 License Agreement”) with the University of Illinois at Chicago (“UIC”) that replaced a 2007 license agreement with Pier that had been terminated. The 2014 License Agreement grants the Company, among other provisions, exclusive rights: (i) to practice certain patents in the United States, and certain other countries as set forth in the 2014 License Agreement; (ii) to identify, develop, make, have made, import, export, lease, sell, have sold or offer for sale any related licensed products; and (iii) to grant sub-licenses of the rights granted in the 2014 License Agreement, subject to the provisions of the 2014 License Agreement. The 2014 License Agreement obligates the Company to pay UIC a license fee, royalties, patent costs and certain milestones. Royalty payments include a royalty on net sales of 4%, payment on sub-licensee revenues of 12.5%, and a minimum annual royalty beginning in 2015 of $100,000, which is due and payable on December 31 of each year beginning on December 31, 2015. The due date of the minimum annual royalty obligation of $100,000 originally due on December 31, 2020, was extended to April 19, 2021 and was paid on April 1, 2021. A one-time milestone payment will be due within 5 days of any of the following, (a) dosing of the first patient with a dronabinol product in a Phase 2 human clinical study anywhere in the world that is not sponsored by the University of Illinois, (b) dosing of the first patient in a Phase 2 human clinical study anywhere in the world with a low dose dronabinol (defined as less than or equal to 1 mg), or (c) dosing of the first patient in a Phase 1 human clinical study anywhere in the world with a proprietary reformulation of dronabinol. One-time milestone payments may become due based upon the achievement of certain development milestones. $350,000 will be due within five days after the dosing of the first patient in a Phase 3 human clinical trial anywhere in the world. $500,000 will be due within five days after the first NDA filing with the FDA, as defined below, or a foreign equivalent. $1,000,000 will be due within twelve months of the first commercial sale. One-time and annual royalty payments may also become due and payable. In the year after the first application for market approval is submitted to the FDA or a foreign equivalent and until approval is obtained, the minimum annual royalty will increase to $150,000. In the year after the first market approval is obtained from the FDA or a foreign equivalent and until the first sale of a product, the minimum annual royalty will increase to $200,000. In the year after the first commercial sale of a product, the minimum annual royalty will increase to $250,000. For each of the fiscal years ended December 31, 2020 and 2019, the Company recorded a charge to operations of $100,000 as its minimum annual royalty obligation, which is included in research and development expenses in the Company’s consolidated statements of operations for the fiscal years ended December 31, 2020 and 2019, respectively.
RespireRx has exclusive rights to issued and pending patents claiming cannabinoid compositions and methods for treating cannabinoid-sensitive disorders, including sleep apnea, pain, glaucoma, muscular spasticity, anorexia and other conditions. In October 2019, we filed a continuation-in-part for our pending patent that describes and claims novel doses, controlled release compositions and methods of use for cannabinoids, and in January 2021, a provisional patent application further disclosing novel dosage and controlled release compositions and methods of use for cannabinoids, alone or in combination, including with cannabinoid and non-cannabinoid molecules. Specific claims describe low dosage strengths and controlled release formulations for attaining a therapeutic window of cannabinoid blood levels that produce the desired therapeutic effect(s) for a controlled period of time, while minimizing undesirable side effects. Certain original patents were filed by RespireRx and are now included in the 2014 License Agreement. The subject matter of the provisional patent application filed in January 2021 is still available to be filed internationally. See Note 9. Commitments and Contingencies—University of Illinois 2014 Exclusive License Agreement in the notes to our consolidated financial statements as of December 31, 2020, included in this Offering Statement and in our 2020 Form 10-K for more information on the 2014 License Agreement. While no assurance can be provided that the claims in this continuation-in-part or the U.S. provisional patent application will be allowed in whole or in part, or that the patents will ultimately issue, we believe that these new filings, if allowed, will provide market protections through January 2042.
We believe our intellectual property initiatives may afford expanding strategic options and market exclusivity in the burgeoning pharmaceutical cannabinoid business sector. New cannabinoid formulation technology, including nano- and micro-emulsions and thin films, have been shown to bypass the normal route of absorption and liver metabolism of cannabinoids, thus dramatically increasing blood levels and allowing for the use of low doses. Similarly, technologies may be used to achieve a controlled release of dronabinol, and we believe that our pending patent priority relating back to 2010 predates the efforts of others seeking to develop low-dose or controlled or extended release formulations of cannabinoids.
| 25 |
Data from our Phase 2 clinical trials has allowed us to design new proprietary formulations of dronabinol, disclosed in our patent filings and optimized for the treatment of not only OSA, but also other indications. In support of this formulation program, David Dickason joined the Company as Senior Vice-President Preclinical Product Development on September 15, 2020. Mr. Dickason has an extensive background in product formulation development. In laboratory studies, he has generated data confirming the potential for the creation of a proprietary dronabinol formulation which should allow for optimized dose and duration of action for treating OSA. If successful in our development efforts, we believe that the development of a proprietary formulation of dronabinol for RespireRx based on our pending patents for low-dose and extended release dronabinol could lead to the development of a marketable proprietary formulation of dronabinol. We also believe that the development of a novel, proprietary formulation of dronabinol would only extend time to market entry by approximately 12 months compared to the market entry with a currently available generic soft gel capsule but would increase market value and extend market exclusivity given the anticipated new patents and our belief that certain strategic partners would be more interested in a proprietary formulation than an existing one; however, no assurance can be provided that any of the formulation technologies that we are currently analyzing will result in viable products.
Proposed Regulatory Approach for Dronabinol
In conjunction with its management and consultants, the Company intends to file a new NDA under Section 505(b)(2) of the Federal Food, Drug and Cosmetic Act (as amended, the “FDCA” and such NDA a “505(b)(2) NDA”), claiming the efficacy and safety of our proposed proprietary dronabinol formulation in the treatment of OSA. We believe the use of dronabinol for the treatment of OSA is a novel indication for an already approved drug, making it eligible for a 505(b)(2) NDA, as opposed to the submission and approval of a full 505(b)(1) NDA.
The 505(b)(2) NDA was created by the Hatch-Waxman Act, as amended (the “Hatch-Waxman Act”), which amended the FDCA to help avoid unnecessary duplication of studies already performed on a previously approved drug. As amended, the FDCA gives the FDA express permission to rely on data not developed by the NDA applicant. Accordingly, a 505(b)(2) NDA contains full safety and effectiveness reports but allows at least some of the information required for NDA approval, such as safety and efficacy information about the active ingredient, to come from studies not conducted by or for the applicant. This can result in a less expensive and faster route to approval, compared with a traditional development path, such as 505(b)(1), while still allowing for the creation of new, differentiated products. The 505(b)(2) NDA regulatory path offers the applicant market protections, such as market exclusivity, under the Hatch-Waxman Act and the rules promulgated thereunder. Other, international regulatory routes are available to pursue proprietary formulations of dronabinol and would provide further market protections. For example, in Europe, a regulatory approval route similar to the 505(b)(2) pathway is the hybrid procedure based on Article 10 of Directive 2001/83/EC.
We have worked with regulatory consultants who will assist with FDA filings and regulatory strategy. If we can secure sufficient financing, of which no assurance can be provided, we anticipate requesting a pre-IND (pre-Investigational New Drug application) meeting with the FDA. This meeting also could create the type of dialogue with the FDA that is normally communicated at an end-of-Phase 2 meeting. The FDA responses to this meeting will be incorporated into an IND.
If we can secure sufficient financing and successfully create a proprietary formulation of dronabinol, of which no assurance can be provided, we plan to propose conducting the appropriate clinical studies with our proprietary controlled release formulation in OSA patients to determine safety, pharmacokinetics (“PK”) and efficacy, as well as a standard Phase 1 clinical study to determine potential abuse liability. When a Phase 3 study is required for a 505(b)(2), usually only one study with fewer patients is necessary versus the two, large scale, confirmatory studies generally required for the standard 505(b)(1) NDA. While no assurance can be provided, with an extensive safety database tracking chronic, long-term use of Marinol® and generics, we believe that the FDA should not have major safety concerns with dronabinol in the treatment of OSA.
The Company has worked with the investigators who conducted the Phase 2B clinical trial and our Clinical Advisory Panel to design a draft Phase 3 protocol that, based on the experience and results from the Phase 2A and Phase 2B trials, we believe will provide sufficient data for FDA approval of a RespireRx dronabinol controlled release formulation for OSA. The current version of the protocol is designed as a 90-day randomized, blinded, placebo-controlled study of dronabinol in the treatment of OSA. Depending on feedback from the FDA, the Company estimates that the Phase 3 trial would require between 120 and 300 patients at 15 to 20 sites, and take 18 to 24 months to complete, at a cost of between $10 million and $14 million.
We believe our rights under the Purisys Agreement would help facilitate regulatory approval. See “Business—Manufacturing” in this Offering Circular for information on the Purisys Agreement. Under the Purisys Agreement, Purisys has agreed to (i) provide all of the API estimated to be needed for the clinical development process for first- and second-generation products, three validation batches for NDA filings and adequate supply for the initial inventory stocking for the wholesale and retail channels, subject to certain limitations, (ii) maintain or file valid DMFs with the FDA or any other regulatory authority and provide the Company with access or a right of reference letter entitling the Company to make continuing reference to the DMFs during the term of the agreement in connection with any regulatory filings made with the FDA by the Company, (iii) participate on a development committee, and (iv) make available its regulatory consultants, collaborate with any regulatory consulting firms engaged by the Company and participate in all FDA or DEA meetings as appropriate and as related to the API.
| 26 |
In consideration for these supplies and services, the Company has agreed to (i) purchase exclusively from Purisys, during the commercialization phase, all API for these products at a pre-determined price subject to certain producer price adjustments and (ii) allow Purisys’s participation in the economic success of the commercialized products up to the earlier of the achievement of a maximum dollar amount or the expiration of a period of time.
Large Commercial Opportunity
As a serious public health issue, the important need for diagnosing and ultimately treating OSA has recently been highlighted by the FDA clearance of several sleep apnea home test kits that are now third party reimbursed. Further highlighting this need, CVS Health Corporation (NYSE: CVS) announced the implementation of a program to diagnose and treat OSA initially within its own in-store, walk-in MinuteClinics. If implemented throughout its HealthHUB store network, we expect the number of people diagnosed with sleep apnea and eligible for treatment to increase dramatically. Fitbit, Inc., (NYSE: FIT), a health oriented smart watch company is seeking clearance from the FDA to diagnose sleep apnea using its smart watches. We believe that the combination of more efficient and patient friendly diagnostic procedures and, ultimately, pharmaceutical treatments such as those we are developing will encourage more patients to seek diagnosis and treatment. As noted above, there are approximately 29 million OSA patients in the United States and an additional 26 million in Germany and 8 million in the United Kingdom. There are currently no drugs approved for the treatment of OSA.
EndeavourRx – Neuromodulators
Background
As described above, during the neurotransmission process, neurons release neurotransmitters that attach to specific receptors residing on adjacent neurons, enabling them to communicate with one another and produce excitatory or inhibitory effects. For example, glutamate is the primary excitatory neurotransmitter in the brain and GABA is the primary inhibitory neurotransmitter. While the neurotransmitter attachment site on each of these receptors does not change, the receptor protein subunit structures can vary so that the receptors can produce a variety of effects. With the AMPA glutamate receptor, the binding of glutamate or an artificial agonist to its attachment site causes a change in the structure of the AMPA receptor resulting in an influx of cations and an increased excitability. Likewise, in the case of the GABAA receptor, the binding of GABA or an artificial agonist to its attachment site causes a change in the structure of the GABAA receptor ion channel and increases the flow of chloride ions (negatively charged anion) into the cell, resulting in decreased excitability.
Neurotransmitter receptor proteins also may contain auxiliary “allosteric” binding sites, which are located adjacent to the agonist binding sites at which neurotransmitters act. Unlike neurotransmitters, neuromodulators are drugs that act at these allosteric binding sites rather than directly at the agonist binding site. They can act either as PAMs, which enhance, or as negative allosteric modulators (“NAMs”), which reduce, the actions of neurotransmitters at their primary receptor sites. Neuromodulators have no intrinsic activity of their own. We have coined the terms “AMPAkines” and “GABAkines” to refer to drugs that act as PAMs at the AMPA and GABAA receptors, respectively. By enhancing the effects of neurotransmitters without altering the normal pattern of neuronal activity, neuromodulators offer the possibility of developing “kinder and gentler” neuropharmacological drugs effective in certain neurological and neuropsychiatric disorders, with greater pharmacological specificity and reduced side effects.
Proposed Regulatory Approach for AMPAkines and GABAkines
In conjunction with its management and consultants, the Company intends to initially perform appropriate and required preclinical studies with its GABAkines and file investigational new drug applications (“INDs”) to commence clinical trials with one or more of those drug candidates and either amend existing INDs or file new INDs for its AMPAkines in order to conduct additional clinical trials with those drug candidates. If such studies safely show statistically significant improvement in appropriate clinical endpoints they would likely result in the filing of one or more NDAs for the AMPAkine(s) under Section 505(b)(1) of the Federal Food, Drug and Cosmetic Act as amended, the traditional regulatory path for new chemical entities (NCEs). The NDAs for the GABAkine drug product candidates, also would be filed as 505(b)(1) NDAs.
As part of our effort to capitalize upon a possible market opportunity with respect to neuromodulators, the Company has implemented an internal restructuring plan, by which EndeavourRx became a stand-alone business unit focused on the neuromodulator market. EndeavourRx comprises our AMPAkine program and our GABAkine program.
AMPAkines
The Company is developing a class of proprietary compounds known as AMPAkines, which are PAMs of the AMPA glutamate receptor. AMPAkines are small molecule compounds that enhance the excitatory actions of glutamate at the AMPA receptor complex, which mediates most excitatory transmission in the central nervous system (“CNS”). Through an extensive translational research effort from the cellular level through Phase 2 clinical trials, we have developed a family of AMPAkines, including CX717, CX1739 and CX1942 that may have clinical application in the treatment of CNS-driven neurobehavioral and cognitive disorders, Spinal Cord Injury (“SCI”), neurological diseases, and certain orphan indications. CX717 and CX1739, our lead clinical compounds, have successfully completed multiple Phase 1 safety trials with no drug-associated serious adverse events. Both compounds have also completed Phase 2 efficacy trials demonstrating target engagement, by antagonizing the process of opioid-induced respiratory depression (“OIRD”). CX717 has successfully completed a Phase 2 trial demonstrating the ability to significantly reduce the symptoms of adult ADHD. In an early Phase 2 study, CX1739 improved breathing in patients with central sleep apnea. In addition, preclinical studies have highlighted the potential ability of these AMPAkines to improve motor function in animals with SCI. Subject to raising sufficient financing (of which no assurance can be provided), we believe that we will be able to initiate a human Phase 2 study with CX1739 or CX717 in patients with SCI and a human Phase 2 study in patients with ADHD using either CX1739 or CX717.
| 27 |
AMPAkines as Treatment for ADHD
ADHD is a relatively common neurobehavioral disorder. Currently available treatments for ADHD include amphetamine-type stimulants and non-stimulant agents targeting monoaminergic neurotransmitter systems in the brain. However, these neurotransmitter systems are not restricted to the brain and are widely found throughout the body. Thus, while these agents can be effective in ameliorating ADHD symptoms, they also can produce adverse cardiovascular effects, such as increased heart rate and blood pressure. Existing treatments also affect eating habits and can reduce weight gain and growth in children and have been associated with suicidal ideation in adolescents and adults. In addition, approved stimulant treatments are DEA classified as controlled substances and present logistical issues for distribution and protection from diversion. Approved non-stimulant treatments, such as atomoxetine (Strattera® and its generic equivalents), can take four to eight weeks to become effective and undesirable side effects also have been observed.
Various investigators have generated data supporting the concept that alterations in AMPA receptor function might underlie the production of some of the symptoms of ADHD. In rodent and primate models of cognition, AMPAkines have been demonstrated to reduce inattention and impulsivity, two of the cardinal symptoms of ADHD. Furthermore, AMPAkines do not stimulate spontaneous locomotor activity in either mice or rats, unlike the stimulants presently used for the treatment of ADHD, nor do they increase the stimulation produced by amphetamine or cocaine. These preclinical considerations prompted us to conduct a randomized, double-blind, placebo controlled, two period crossover study to assess the efficacy and safety of CX717 in adults with ADHD.
In a repeated measures analysis, a statistically significant treatment effect on the ADHD Rating Scale (ADHD-RS), the primary outcome measure, was observed after a three-week administration of CX717, at a dose of 800 mg BID. Differences between this dose of CX717 and placebo were observed as early as week one of treatment and continued throughout the remainder of the study. The low dose of CX717, 200 mg BID, did not differ from placebo. In general, results from both the ADHD-RS hyperactivity and inattentiveness subscales, which were secondary efficacy variables, paralleled the results of the total score. CX717 was considered safe and well tolerated.
Based on these clinical results, we believe that AMPAkines such as CX717 or CX1739 might represent a breakthrough opportunity to develop a non-stimulating therapeutic for ADHD with the rapidity of onset normally seen with stimulants. Subject to raising sufficient financing (of which no assurance can be provided), we are planning to continue this program with a Phase 2 clinical trial in patients with adult ADHD using one of our two lead ampakine compounds.
AMPAkines as Treatment for SCI
AMPAkines also may have potential utility in the treatment and management of SCI to enhance motor functions and improve the quality of life for SCI patients. An estimated 17,000 new cases of SCI occur each year in the United States, most a result of automobile accidents. Currently, there are roughly 282,000 people living with spinal cord injuries, which often produce impaired motor function.
SCI can profoundly impair neural plasticity leading to significant morbidity and mortality in human accident victims. Plasticity is a fundamental property of the nervous system that enables continuous alteration of neural pathways and synapses in response to experience or injury. A large body of literature exists regarding the ability of AMPAkines to stimulate neural plasticity, possibly due to an enhanced synthesis and secretion of various growth factors.
The Company has been working with Dr. David Fuller at the University of Florida which has funding from NIH, to evaluate the use of AMPAkines for the treatment of compromised motor function in SCI. Using mice that have received spinal hemi-sections, CX717 was observed to increase motor nerve activity bilaterally. The effect on the hemisected side was greater than that measured on the intact side, with the recovery approximating that seen on the intact side prior to administration of ampakine. The doses of AMPAkines active in SCI were comparable to those demonstrating antagonism of OIRD, indicating target engagement of the AMPA receptors.
Recently, studies in patients with SCI have demonstrated that neural plasticity can be induced to improve motor function. This is based on the ability of spinal circuitry to learn how to adjust spinal and brainstem synaptic strength following repeated hypoxic bouts. Animal studies have demonstrated the ability of AMPAkines to dramatically enhance the effects of AIH on motor neuron activity after SCI. Because AMPAkines are known to enhance synaptic plasticity, the potential exists to harness repetitive AIH in combination with AMPAkines as a means of inducing functional recovery of motor function following SCI.
| 28 |
These animal models of motor nerve function following SCI support proof of concept for a new treatment paradigm using AMPAkines to improve motor functions in patients with SCI. With additional funding granted by NIH to Dr. Fuller, the Company is continuing its collaborative preclinical research with him, while it is planning a clinical trial program focused on developing AMPAkines for the restoration of certain motor functions in patients with SCI. The Company is working with researchers at highly regarded clinical sites to finalize a Phase 2 clinical trial protocol. We believe that a clinical study could be initiated within several months of raising sufficient financing (of which no assurance can be provided).
GABAkines
The GABAkine program was established pursuant to the UWMRF Patent License Agreement. At present, the program is focused on developing novel GABAkines with certain GABAA receptor subtype selectivity. We believe that there is a considerable degree of receptor subtype heterogeneity, making subtype selectivity of our compounds a desirable attribute.
On August 1, 2020, RespireRx exercised its option pursuant to its option agreement dated March 2, 2020, between RespireRx and UWMRF. Upon exercise RespireRx and UWMRF executed the UWMRF Patent License Agreement effective August 1, 2020 (the “Effective Date”) pursuant to which RespireRx licensed the identified intellectual property. Under the UWMRF Patent License Agreement, the Company has an exclusive license to commercialize GABAkine products based on UWMRF’s rights in certain patents and patent applications, and a non-exclusive license to commercialize products based on UWMRF’s rights in certain technology that is not the subject of the patents or patent applications. UWMRF maintains the right to use, and, upon the approval of the Company, to license, these patent and technology rights for any non-commercial purpose, including research and education. The UWMRF Patent License Agreement expires upon the later of the expiration of the Company’s payment obligations to UWMRF or the expiration of the last remaining licensed patent granted thereunder, subject to early termination upon the occurrence of certain events. The License Agreement also contains a standard indemnification provision in favor of UWMRF and confidentiality provisions obligating both parties. Under the UWMRF Patent License Agreement, in consideration for the licenses granted, the Company will pay to UWMRF the following: (i) patent filing and prosecution costs incurred by UWMRF prior to the Effective Date, paid in yearly installments over three years from the Effective Date; (ii) annual maintenance fees, beginning on the second anniversary of the Effective Date, ranging from $5,000 on the second anniversary to $15,000 on the fifth anniversary and each anniversary thereafter, which annual maintenance fees terminate upon the Company’s payment of royalties pursuant to clause (iv) below; (iii) milestone payments, paid upon the occurrence of certain dosing events of patients during clinical trials and certain approvals by the FDA, such milestone payments not to exceed $2,150,000 in the aggregate; and (iv) royalties on net sales of products developed with the licenses, subject to minimum annual payments and to royalty rate adjustments based on whether separate royalty payments by the Company yield an aggregate rate beyond a stated threshold. The Company has also granted UWMRF certain stock appreciation rights with respect to the Company’s neuromodulator programs, subject to certain limitations, and will pay to UWMRF certain percentages of revenues generated from sublicenses of the licenses provided under the License Agreement by the Company to third parties.
Benzodiazepines (“BDZs”), such as Valium® (diazepam), Librium® (chlordiazepoxide) and Xanax® (alprazolam) were the first major class of drugs reported to act as GABAA PAMs, by binding at a site distinct from the binding site for GABA. These drugs produce a wide range of pharmacological properties, including anxiety reduction, sedation, hypnosis, anti-convulsant, muscle relaxation, respiratory depression, cognitive impairment, as well as tolerance, abuse and withdrawal. For this reason, it was not surprising that BDZs were observed to act as GABAA PAMs indiscriminately across all GABAA receptor subtypes. Following the identification of BDZ binding sites on GABAA receptors, Dr. Lippa described CL218,872, the first non-BDZ to demonstrate that these receptors were heterogeneous by binding selectively to a subtype of GABAA receptor. This demonstration of receptor heterogeneity led to the hypothesis that the various pharmacological actions of the BDZs might be separable depending on the receptor subtype involved. In animal testing, CL218,872 provided the proof of principle that such a separation could be achieved by displaying anti-anxiety and anti-convulsant properties in the absence of sedation, amnesia and muscular incoordination. Using ocinaplon, an analog of CL218,872 with similar receptor subtype selectivity, Dr. Lippa’s team reported in the Proceedings of the National Academy of Science the results of a Phase 2 clinical trial in anxious patients that ocinaplon significantly reduced symptoms of anxiety in the absence of sedation. These findings gave impetus to the search for novel therapeutic drugs for neurological and psychiatric illnesses that display improvements in efficacy and reductions in side effects.
Over the last several years, a group of scientists led by Dr. James Cook of the University of Wisconsin and Dr. Jeffrey Witkin affiliated with the Indiana University School of Medicine, have synthesized and tested a broad series of novel drugs that display GABAA receptor subtype selectivity and pharmacological specificity. Certain of these chemical compounds are the subject of the UWMRF Patent License Agreement and Drs Cook and Witkin have been engaged as consulting Research Fellows, while still maintaining their academic affiliations.
Of these compounds, we have identified KRM-II-81 as a clinical lead. KRM-II-81 is the most advanced and druggable of a series of compounds that display certain receptor subtype selectivity and pharmacological specificity. In studies using cell cultures, brain tissues and whole animals, KRM-II-81 acts as a GABAA PAM at selective GABAA receptor subtypes that we feel are intimately involved in neuronal processes underlying epilepsy, pain, anxiety and certain other indications. KRM-II-81 has demonstrated highly desirable properties in animal models of these and other potential therapeutic indications, in the absence of or with greatly reduced liability to produce sedation, motor incoordination, cognitive impairments, respiratory depression, tolerance, abuse and withdrawal seizures, all side effects associated with BDZs. We currently are focused on the potential treatment of epilepsy and pain.
| 29 |
Epilepsy and Existing Treatments
Epilepsy is a chronic and highly prevalent neurological disorder that affects millions of people world-wide and has serious consequences for the life of the affected individual. A first-line approach to the control of epilepsy is through the administration of anticonvulsant drugs. Repeated, uncontrolled seizures due to drug resistance and the side effects arising from seizure medications have a negative effect on the developing brain and can lead to brain cell loss and severe impairment of neurocognitive function. The continued occurrence of seizure activity also increases the probability of subsequent epileptic events through sensitization mechanisms called seizure kindling. Seizures that are unresponsive to anti-epileptic treatments are life-disrupting and life-threatening with broad health, life, and economic consequences.
Like many diseases, epilepsy is still remarkably underserved by currently available medicines. Pharmaco-resistance to anticonvulsant therapy continues to be one of the key obstacles to the treatment of epilepsy. Although many anticonvulsant drugs are approved to decrease seizure probability, seizures frequently are not fully controlled and patients are generally maintained daily on multiple antiepileptic drugs with the hope of enhancing the probability of seizure control. Despite this polypharmacy approach, as many as 60% to 70% of patients continue to have seizures. As a result of the lack of seizure control, pharmaco-resistant epilepsy patients, including young children, sometimes require and elect to have invasive therapeutic procedures such as surgical resection of targeted brain tissue.
Despite the availability of a host of marketed drugs of different mechanistic classes, the lack of seizure control in patients is the primary factor driving the need for improved antiepileptic drugs, as emphasized by researchers and patient advocacy communities. Increasing inhibitory tone in the CNS through enhancement of GABAergic inhibition is a proven mechanism for seizure control. However, GABAergic medications also exhibit liabilities that limit their antiepileptic potential. Tolerance develops to GABAergic drugs such as BDZs, limiting their use in a chronic setting. These drugs can produce cognitive impairment, somnolence, sedation, tolerance and withdrawal seizures that create dosing limitations such that they are generally used only for acute convulsive episodes.
GABAkines as Treatments for Epilepsy
KRM-II-81 has demonstrated efficacy in multiple rodent models and measures of antiepileptic drug efficacy in vivo. This includes nine acute seizure provocation models in mice and rats, four seizure sensitization models in rats and mice, two models of chronic epilepsy, and three models specifically testing pharmaco-resistant antiepileptic drug efficacy. Because it appears to have a substantially reduced side effect liability, it might be possible to use higher, more effective doses than standard of care medications. Predictions of superior efficacy of KRM-II-81 over standard of care anti-epileptics comes from the efficacy of this compound across a broad range of animal models of epilepsy. Importantly, KRM-II-81 has been shown to be effective in models assessing pharmaco-resistant epilepsy. Under these conditions, KRM-II-81 is efficacious in cases where standard of care medicines do not work.
In the absence of seizure control by anti-epileptics, surgical resection of affected brain tissue is one potential alternative to help with the control of seizures. In the process of this surgery, epileptic brain tissue can become available for research into epileptic mechanisms and the identification of novel antiepileptic drugs. The anticonvulsant action of KRM-II-81 has been confirmed by microelectrode recordings from slices obtained from freshly excised cortex tissue from epileptic patients where in situ application of KRM-II-81 suppressed epileptiform electrical activity. While preliminary, these translational data lend support to the further development of KRM-II-81 for the treatment of epilepsy.
GABAkines as Treatments for Pain
It is impossible not to be aware of the crisis that the opioid epidemic has created in the treatment of chronic pain. While there is no question as to their efficacy, the clinical use of opioids is severely limited due to the rapid development of tolerance, dependence and the production of OIRD, the major cause of opioid-induced lethality. Research programs are underway nationwide to discover and develop new non-opioid drugs that are effective analgesics without the tolerance and abuse liability ascribed to opioids. Chronic pain is especially difficult to treat due to its complex nature with a variety of different etiologies. For example, chronic pain may be produced by injury, surgery, neuropathy, the inflammation produced by arthritis or by certain drugs such as cancer chemotherapeutics. For these reasons, better management and control of chronic pain continues to be a serious need in medical practice.
Data from both preclinical and clinical studies are consistent with the idea that GABAergic neurotransmission is an important regulatory mechanism for the control of pain. Gabapentin (Neurontin®) and pregabalin (Lyrica®), two commonly used drugs for the treatment of chronic pain, are believed to produce their analgesic effects by enhancing GABAergic neurotransmission. However, although they have received FDA approval, the clinical results have not been overwhelming. In a published review of 37 clinical trials with a total of 5,914 patients experiencing neuropathic pain there was no difference in the percentage of patients experiencing pain reduction of greater than 50% when comparing gabapentin to placebo. The most common side effects produced by gabapentin were sedation, dizziness and problems walking. It is uncertain whether greater efficacy was not observed because of poor intrinsic pharmacological efficacy or insufficient dosages due to dose limiting side effects.
| 30 |
An alternate approach to enhancing GABAergic neurotransmission is the use of GABAA PAMs. This approach has been under-utilized because of the general lack of efficacy of the BDZ PAMs. However, a strong case for the potential value of subtype selective GABAA PAMs for the treatment of pain can be made. First, GABAA receptor regulated pathways are integral to pain processing with α2/3 containing GABAA receptor subtypes present on nerve pathways modulating pain sensation and perception. Second, we believe that the analgesic properties of BDZs may be masked by concurrent activation of other GABAA receptor subtypes that mediate the side effects. Diazepam has been reported to produce maximal analgesia in rodents if the side effects are attenuated by GABAA subtype genetic manipulation. Third, KRM-II-81 and predecessor GABAkines made by Dr. Cook, which selectively amplify GABAA receptor subtype signaling, are effective in pain models in rodents at doses much lower than those producing motor side effects.
In a number of laboratory procedures and animal studies, KRM-II-81 has been shown to selectively bind to GABAA receptor subtypes and enhance GABAergic neurotransmission. Sub-chronic dosing for 22 days with KRM-II-81 and the structural analogue, MP-III-80, demonstrated enduring analgesic efficacy without tolerance development. In contrast, tolerance developed to the analgesic effects of gabapentin. At a dose that produces maximal analgesic effect in an inflammatory chronic pain model, KRM-II-81 does not substitute for the BDZ midazolam in a drug discrimination assay, suggesting a reduced abuse liability. Furthermore, KRM-II-81 did not produce the respiratory depression observed with alprazolam, a major problem with BDZs leading to emergency room visits and overdose.
We believe that the ability to attenuate both acute and chronic pain combined with a greatly reduced side effect profile, a lack of tolerance and a reduced abuse potential makes KRM-II-81 a promising clinical lead and a potential advance in pain therapeutics. Results from preliminary chemistry, metabolism and pharmacokinetic studies support its further development.
Corporate and Product Development Plans
As discussed above, in order to facilitate our business activities and product development, we have organized our drug platforms into two separate business units which currently operate as divisions, but which are anticipated to be re-organized as separate legal entity subsidiaries in the future. ResolutionRx is focused on pharmaceutical cannabinoids and EndeavourRx is focused on neuromodulators. Below is a description of the Company’s product development plans within these business units. Indications of use of proceeds from this Offering described below assume the sale of all 375,000,000 Shares in this Offering at the offering price of $0.02 per share, of which no assurance can be provided. Please see “Use of Proceeds” elsewhere in this Offering Circular for indications of use of proceeds assuming the sale of less than 375,000,000 Shares in this Offering.
We anticipate allocating $1,300,092 of the net proceeds of this Offering for research and development expenditures that are shared across the dronabinol, AMPAkines, and GABAkines programs. Specific additional expenditures within each of these programs is described below.
ResolutionRx – Dronabinol program
The dronabinol program within our ResolutionRx cannabinoid platform is anticipated to directly utilize a total of $400,000 of the net proceeds of this Offering on the continued development of a proprietary formulation of dronabinol. In conjunction with a sub-contractor, the Company already has prepared several new proprietary formulations of dronabinol with the anticipated properties described in our patent applications. The funds from this Offering are planned to be used to complete final evaluation of those versions of dronabinol formulations with the optimum physico-chemical properties and to test them in animal pharmacokinetic studies. Assuming sufficient additional financing is available, of which no assurance can be provided, we intend to engage regulatory consultants, make expenditures for the initial stocking of clinical supply, packaging and distribution in anticipation of performing Phase 2 PK and PD (pharmacodynamic) clinical trials and ultimately one or more pivotal Phase 3 clinical studies.
The Purisys Agreement and the 2014 License Agreement will need to be transferred or otherwise made available to ResolutionRx. See “—Noramco Inc./Purisys, LLC - Dronabinol Development and Supply Agreement” and “—University of Illinois 2014 Exclusive License Agreement” in Note 9. Commitments and Contingencies in the notes to consolidated financial statements as of December 31, 2020 in this Offering Statement and in our 2020 Form 10-K for more information on these agreements. Initially, ResolutionRx’s primary focus will be on re-purposing dronabinol for the treatment of OSA; we believe that our broad enabling patents and our 2019 and 2021 patent applications for proprietary formulation technology may provide a framework for expanding into the larger burgeoning pharmaceutical cannabinoid industry. We believe that by converting this division to a subsidiary, it may be possible, through separate finance channels and potential strategic transactions, to optimize the asset value not only of the cannabinoid platform, but separately, our neuromodulator platform as well.
EndeavourRx – AMPAkines program
For the AMPAkines program within our EndeavourRx neuromodulators platform, the Company plans to directly utilize $1,525,000 of the net proceeds of the Offering to assess the purity of our existing drug supplies, obtain clinical supply material, engage regulatory consultants and a contract research organization (CRO) to finalize a clinical trial protocol and conduct a Phase 2A clinical trial to determine the safety, pharmacokinetic (PK) and pharmacodynamic (PD) properties of one of our lead AMPAkines in patients who have had SCI. These tasks are critical for applying to the FDA for permission to amend our existing IND or initiate a new IND enabling the commencement of clinical trials.
Assuming sufficient additional financing is available, of which no assurance can be provided, the Company would continue to focus on SCI, and in particular, a Phase 2A efficacy study, as we believe it would be the most efficient expenditure of our resources and yield an actionable result in the shortest period of time, and would initiate additional clinical trials in patients with ADHD.
| 31 |
EndeavourRx – GABAkines program
For the GABAkines program, the Company plans to directly utilize $650,000 of the net proceeds of the Offering to obtain active pharmaceutical ingredient and have it quality control tested and conduct animal toxicity studies.
Assuming sufficient additional financing in available, of which no assurance can be provided, the Company would conduct a full preclinical program in anticipation of filing an IND to commence human clinical trials for safety and efficacy in patients with treatment resistant epilepsy and those requiring non-opioid treatments for pain.
In connection with the organization and development of the ResolutionRx and EndeavourRx business units, we are planning certain corporate and development actions as summarized below. All of the below are subject to raising additional financing and/or entering into strategic relationships, of which no assurance can be given.
Proposed Creation of Subsidiaries
Pending approval by the Board of Directors, management intends to organize our ResolutionRx and EndeavourRx business units into two subsidiaries: (i) a ResolutionRx subsidiary, into which we intend to contribute our pharmaceutical cannabinoid platform and its related tangible and intangible assets and certain of its liabilities and (ii) an EndeavourRx subsidiary, into which we plan to contribute our neuromodulator platform, including both the AMPAkine and GABAkine programs and their related tangible and intangible assets and certain of their liabilities.
Management believes that there are several advantages to separating these platforms formally into newly formed subsidiaries, including but not limited to optimizing their asset values through separate finance channels and making them more attractive for capital raising as well as for strategic deal making.
Employee/Consultant Infrastructure Build-out
It is anticipated that the Company will continue to use, at least initially, its management personnel to provide management, operational and oversight services to these two business units. In order to broaden our operational expertise, we are planning to hire a number of highly qualified individuals, either as employees or consultants and, in tandem, increase our administrative support function. To date, we have hired David Dickason as Senior Vice-president of Pre-Clinical Product Development and engaged Drs. James Cook and Jeffrey Witkin as consulting Research Fellows and engaged Dr. Rok Cerne as Senior Research Scientist.
Competition
The pharmaceutical industry is characterized by intensive research efforts, rapidly advancing technologies, intense competition and a strong emphasis on proprietary therapeutics. Our competitors include many companies, research institutes and universities that are working in a number of pharmaceutical or biotechnology disciplines to develop therapeutic products similar to those we are currently investigating. Most of these competitors have substantially greater financial, technical, manufacturing, marketing, distribution and/or other resources than we do. In addition, many of our competitors have experience in performing human clinical trials of new or improved therapeutic products and obtaining approvals from the FDA and other regulatory agencies. We have no experience in conducting and managing later-stage clinical testing or in preparing applications necessary to obtain regulatory approvals. We expect that competition in this field will continue to intensify.
Regulatory Requirements for Drug Market Approval
The FDA and other similar agencies in foreign countries have substantial requirements for therapeutic products. Such requirements often involve lengthy and detailed laboratory, clinical and post-clinical testing procedures and are expensive to complete. It often takes companies many years to satisfy these requirements, depending on the complexity and novelty of the product. The review process is also extensive, which may delay the approval process further. Failure to comply with applicable FDA or other requirements may subject a company to a variety of administrative or judicial sanctions, such as the FDA’s refusal to approve pending applications, a clinical hold, warning letters, recall or seizure of products, partial or total suspension of production, withdrawal of the product from the market, injunctions, fines, civil penalties or criminal prosecution.
FDA approval is required before any new drug or dosage form, including the new use of a previously approved drug, can be marketed in the United States. Other similar agencies in foreign countries also impose substantial requirements.
| 32 |
The process of developing drug candidates normally begins with a discovery process of potential candidates that are then initially tested in in vitro and in vivo non-human animal (preclinical) studies which include but are not limited to toxicity and other safety related studies, pharmacokinetics, pharmacodynamics and ADME (absorption, distribution, metabolism, excretion). Once sufficient preclinical data are obtained, a company must submit an IND and receive authorization from the FDA in order to begin clinical trials in the United States. Successful drug candidates then move into human studies that are characterized generally as Phase 1, Phase 2 and Phase 3. Phase 1 studies seeking safety and other data normally utilize healthy volunteers. Phase 2 studies utilize one or more prospective patient populations and are designed to establish safety and preliminary measures of efficacy. Sometimes studies may be referred to as Phase 2A and 2B depending on the size of the patient population. Phase 3 studies are large trials in the targeted patient population, performed in multiple centers, often for longer periods of time and are designed to establish statistically significant efficacy as well as safety in the larger population. Most often the FDA and similar regulatory agencies in other countries require two confirmatory Phase 3 or pivotal studies. Upon completion of both the preclinical and clinical phases, an NDA (New Drug Application) is filed with the FDA or a similar filing is made to the regulatory authority in other countries. NDA filings are extensive and include the data from all prior studies. These filings are reviewed by the FDA and, only if approved, may the company or its partners commence marketing of the new drug in the United States.
There also are variations of these procedures. For example, companies seeking approval for new indications for an already approved drug may choose to pursue an abbreviated approval process such as the filing for an NDA under Section 505(b)(2). Another example would be a Supplementary NDA (“SNDA”). A third example would be an Abbreviated NDA (“ANDA”) claiming bioequivalence to an already approved drug and claiming the same indications such as in the case of generic drugs. Other opportunities allow for accelerated review and approval based upon several factors, including potential fast-track status for serious medical conditions and unmet medical needs, potential breakthrough therapy designation of the drug for serious conditions where preliminary evidence shows that the drug may show substantial improvement over available therapy or orphan designation (generally, an orphan indication in the United States is one with a patient population of less than 200,000).
As of yet, we have not obtained any approvals to market our products. Further, we cannot assure you that the FDA or other regulatory agency will grant us approval for any of our products on a timely basis, if at all. Even if regulatory clearances are obtained, a marketed product is subject to continual review, and later discovery of previously unknown problems may result in restrictions on marketing or withdrawal of the product from the market.
The recent COVID-19 pandemic has made it very difficult to recruit subjects and patients and to conduct clinical trials in general and it is unclear how long these challenges will last. Given the public health emergency during the winter and spring of 2020 which continues into 2021, the FDA issued guidance to be implemented without the normal prior public comment period as the FDA had concluded that public participation would not be feasible or appropriate. Guidance is not legally enforceable, but the FDA recommends the following of its guidance. Challenges are expected to arise from quarantines, site closures, travel limitations, interruptions to the supply chain for investigational products, or other considerations if site personnel or trial subjects become infected with COVID-19. These challenges may lead to difficulties in meeting protocol-specified procedures. The FDA emphasized that safety of trial participants is critically important. Decisions to continue or discontinue individual patients or the trial are expected to be made by trial sponsors in consultation with clinical investors and Institutional Review Boards. COVID-19 screening procedures may need to be implemented. As challenging as the clinical trial process is during normal times, the risks, strategic and operational challenges and the costs of conducting such trials has increased substantially during the pandemic.
See “Risk Factors—Risks related to our business—We may not be able to successfully develop and commercialize our product candidates and technologies.”
Manufacturing
We have no experience or capability to either manufacture bulk quantities of the new compounds that we develop, or to produce finished dosage forms of the compounds, such as tablets or capsules. We rely, and presently intend to continue to rely, on the manufacturing and quality control expertise of contract manufacturing organizations (see below with respect to dronabinol) or current and prospective corporate partners. There is no assurance that we will be able to enter into manufacturing arrangements to produce bulk quantities of our compounds on favorable financial terms. There is generally, absent any disruptions that may be caused by the current pandemic, substantial availability of both bulk chemical manufacturing and dosage form manufacturing capability throughout the world that we believe we can readily access.
On September 4, 2018, RespireRx entered into a dronabinol Development and Supply Agreement with Noramco Inc., one of the world’s major dronabinol manufacturers, which Noramco subsequently assigned to its subsidiary, Purisys LLC. Under the terms of the Purisys Agreement, Noramco agreed to (i) provide all of the active pharmaceutical ingredient (“API”) estimated to be needed for the clinical development process for both the first- and second-generation products (each a “Product” and collectively, the “Products”), three validation batches for New Drug Application (“NDA”) filing(s) and adequate supply for the initial inventory stocking for the wholesale and retail channels, subject to certain limitations, (ii) maintain or file valid drug master files (“DMFs”) with the FDA or any other regulatory authority and provide the Company with access or a right of reference letter entitling the Company to make continuing reference to the DMFs during the term of the agreement in connection with any regulatory filings made with the FDA by the Company, (iii) participate on a development committee, and (iv) make available its regulatory consultants, collaborate with any regulatory consulting firms engaged by the Company and participate in all FDA or Drug Enforcement Agency (“DEA”) meetings as appropriate and as related to the API. We now refer to the second-generation product as our proprietary formulation or proprietary product and have de-emphasized the first-generation product.
| 33 |
In consideration for these supplies and services, the Company has agreed to purchase exclusively from Noramco during the commercialization phase all API for its Products (as defined in the Development and Supply Agreement) at a pre-determined price subject to certain producer price adjustments and agreed to Noramco’s participation in the economic success of the commercialized Product or Products up to the earlier of the achievement of a maximum dollar amount or the expiration of a period of time.
See “Risk Factors—Risks related to our business—We may not be able to successfully develop and commercialize our product candidates and technologies” for a discussion of certain risks related to the development and commercialization of our products.
Marketing
We have no experience in the marketing of pharmaceutical products and do not anticipate having the resources to distribute and broadly market any products that we may develop. We will therefore continue to seek commercial development arrangements with other pharmaceutical companies for our proposed products for those indications that require significant sales forces to effectively market. In entering into such arrangements, we may seek to retain the right to promote or co-promote products for certain of the orphan drug indications in North America. We believe that there is a significant expertise base for such marketing and sales functions within the pharmaceutical industry and expect that we could recruit such expertise if we choose to directly market a drug.
See “Risk Factors—Risks related to our business—We may not be able to successfully develop and commercialize our product candidates and technologies.”
Employees
As of September 30, 2021 and as of the date of filing of this Offering Circular, the Company employed twelve people on a full-time basis. We have five officers, two of whom are part-time outside consultants who are independent contractors, not employees. The Company also engages other contractors who provide substantial services to the Company.
Technology Rights
University of Illinois License Agreement
See ResolutionRx – Pharmaceutical Cannabinoids – The Company’s Cannabinoid Intellectual Property Rights above and see Note 9. Commitments and Contingencies—University of Illinois 2014 Exclusive License Agreement in the notes to our consolidated financial statements as of December 31, 2020 included in this Offering Statement and in our 2020 Form 10-K for more information on the 2014 License Agreement.
UWMRF Patent License Agreement
See EndeavourRx – Neuromodulators – GABAkines above and see Note 9. Commitments and Contingencies—UWMRF Patent License Agreement in the notes to our consolidated financial statements as of December 31, 2020 included in this Offering Statement and in our 2020 Form 10-K for more information on the 2014 License Agreement.
Properties
As of December 31, 2020, the Company did not own any real property or maintain any leases with respect to real property. The Company periodically contracts for services provided at the facilities owned by third parties and may, from time-to-time, have employees who work in these facilities.
Legal Proceedings
We are periodically subject to various pending and threatened legal actions and claims. See Note 9. Commitments and Contingencies – Pending or Threatened Legal Actions and Claims in the notes to our consolidated financial statements for the year ended December 31, 2020 included in this Offering Statement and in our 2020 Form 10-K for additional information regarding these matters.
The legal proceedings discussed in this report could result in adverse judgments, settlements, fines, injunctions, restitutions or other relief that could require significant expenditures or have other effects on our business. Management believes, based on current knowledge and after consultation with counsel, that the outcome of such actions will not have a material adverse effect on our consolidated financial condition. The outcome of litigation and other legal proceedings is inherently uncertain, and it is possible that one or more of the matters currently pending or threatened could have an adverse effect on our liquidity, financial condition or results of operations for any particular period.
| 34 |
Market Price of and Dividends on the Registrant’s Common Equity and Related Stockholder Matters.
Market Information, Holders, and Dividends
Our common stock was quoted on the OTCQB on September 30, 2021 under the symbol “RSPI”. The current quotations on the OTCQB reflect inter-dealer prices, without retail mark-up, mark-down or commission and may not necessarily represent actual transactions.
As of November 26, 2021, there were 124 stockholders of record of our common stock, and approximately 4,000 beneficial owners.
We have never paid cash dividends on our common stock and do not anticipate paying such dividends in the foreseeable future. The payment of dividends, if any, will be determined by the Board in light of conditions then existing, including our financial condition and requirements, future prospects, restrictions in financing agreements, business conditions and other factors deemed relevant by the Board.
During the fiscal year ended December 31, 2020, we did not repurchase any of our securities. During the nine-months ended September 30, 2021, we entered into two warrant exchange agreements pursuant to which we exchanged two previous warrants for two new warrants.
Securities Authorized for Issuance Under Equity Compensation Plans
In March 2014, the Company’s stockholders approved, by written consent, the Cortex Pharmaceuticals, Inc. 2014 Equity, Equity-Linked and Equity Derivative Incentive Plan (“2014 Plan”), filed as exhibit 10.2 to the Company’s Current Report on Form 8-K filed March 24, 2014, which provides for the issuance of shares of Common Stock, in the form of stock grants and options to directors, officers, employees, consultants and other service providers of the Company. There are 32,503 shares authorized and 6,325 available for issuance under the 2014 Plan.
On June 30, 2015, the Board adopted the 2015 Stock and Stock Option Plan (the “2015 Plan”), filed as exhibit 10.1 to the Company’s Current Report on Form 8-K filed July 8, 2015, which similarly provides for the issuance of equity and equity derivative securities such as options. The Company amended the 2015 Plan on March 31, 2016, January 17, 2017, December 9, 2017, December 28, 2018, May 5, 2020, and July 31, 2020 and filed descriptions of such amendments on the Company’s Current Reports on Form 8-K on April 6, 2016, January 23, 2017, December 14, 2017, January 4, 2019, May 6, 2020, and August 3, 2020, respectively. The amendments discussed above primarily increased the number of shares of Common Stock authorized to be issued under the 2015 Plan as approved by the Board, with the August 3, 2020 amendment expanding the number of shares of Common Stock authorized to be issued under the 2015 plan to 158,985,260 shares, which number of shares was adjusted to 15,898,526 upon the consummation of the reverse stock split with respect to Common Stock on January 5, 2021. On July 29, 2021, the Company amended the 2015 Plan to increase the number of shares by an additional 7,000,000 to 22,898,526 shares and filed a description of this amendment on the Company’s Current Report on Form 8-K on July 30, 2021. The Company has not presented, nor does it intend to present, the 2015 Plan, as amended, to shareholders for approval.
The following table sets forth information regarding outstanding options, warrants and rights and shares reserved for future issuance under our existing equity compensation plans as of December 31, 2020 (as adjusted for the reverse stock split).
| Plan Category | Number
of securities to be issued upon exercise of outstanding options, warrants and rights (a) | Weighted average exercise price of outstanding options, warrants and rights (b) | Number
of securities remaining available for issuance under equity compensation plans (excluding securities reflected in column (a)) (c) | |||||||||
| Equity compensation plans approved by security holders | 1,564 | $ | 64.025 | 6,325 | ||||||||
| Equity compensation plans not approved by security holders (including non-plan options) | 7,163,651 | $ | 1.948 | 8,704,251 | ||||||||
| Total | 7,165,215 | $ | 1.961 | 8,710,576 | ||||||||
| 35 |
The table below sets forth information regarding outstanding options, warrants and rights and shares reserved for future issuance under our existing equity compensation plans as of September 30, 2021.
| Plan Category | Number
of securities to be issued upon exercise of outstanding options, warrants and rights (a) | Weighted average exercise price of outstanding options, warrants and rights (b) | Number
of securities remaining available for issuance under equity compensation plans (excluding securities reflected in column (a)) (c) | |||||||||
| Equity compensation plans approved by security holders | 1,564 | $ | 64.025 | 6,325 | ||||||||
| Equity compensation plans not approved by security holders (including non-plan options) | 7,111,343 | $ | 1.419 | 8,756,559 | ||||||||
| Total | 7,112,907 | $ | 1.433 | 8,762,884 | ||||||||
As described above, on July 29, 2021, the Company amended the 2015 Plan to increase the number of shares authorized to be issued under the 2015 Plan by 7,000,000 shares and filed a description of such amendment on the Company’s Current Report on Form 8-K on July 30, 2021. The number of shares authorized to be issued under the 2015 Plan is now 22,898,526, of which 7,109,168 are reserved for issuance pursuant to outstanding equity awards and 15,756,559 remain available for issuance pursuant to awards yet to be granted.
Management’s Discussion and Analysis of Financial Condition and Results of Operations
See Part II. Item 7. Management’s Discussion and Analysis of Financial Condition and Results of Operations in our 2020 Form 10-K and Part I. Item 2. Management’s Discussion and Analysis of Financial Condition and Results of Operations in our Quarterly Report on Form 10-Q for the quarter ended September 30, 2021, as clarified or supplemented by the information below.
Below is a chart that represents our current development status for each of our product candidates for the disorders for which they are being developed. Preclinical testing is pre-human testing and includes in vitro and animal studies. Phase 1 clinical trials are primarily safety, generally conducted in healthy adults. Phase 2 clinical trials are generally somewhat larger than Phase 1 and often include dose finding, additional safety and preliminary efficacy. Phase 3 clinical trials are larger studies designed to test efficacy and safety in a broader population. See Part II. Item 7. Management’s Discussion and Analysis of Financial Condition and Results of Operations in our 2020 Form 10-K for more information on our proposed regulatory approach and development plans for our product candidates.
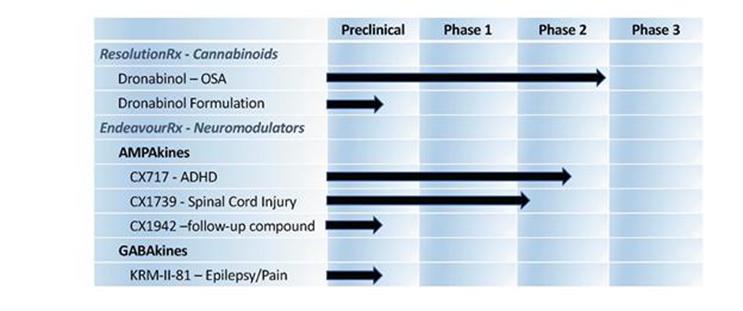
| 36 |
Below is our current business organization with ResolutionRx and EndeavourRx currently operating as divisions and which are planned to become, initially, wholly owned subsidiaries.
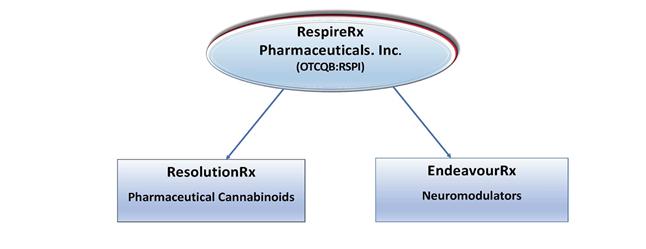
Liquidity and Capital Resources
At September 30, 2021 and December 31, 2020, the Company had cash of $556 and $825, respectively reflecting a decrease in cash of $269 for the nine-months ended September 30, 2021.
The Company is currently, and has for some time, been in significant financial distress. It has limited cash resources and current assets and has no ongoing source of sustainable revenue. Management is continuing to address various aspects of the Company’s operations and obligations, including, without limitation, debt obligations, financing requirements, intellectual property, licensing agreements, legal and patent matters and regulatory compliance, and has continued to raise new debt and equity capital to fund the Company’s general and administrative and research and development activities from both related and unrelated parties. On March 22, 2020, the Company received forgiveness of an aggregate liability of $306,000 in accrued compensation and benefits as consideration for the issuance of 900,000 shares of Common Stock. On July 13, 2020 and September 30, 2020, the Company received forgiveness of an aggregate liability of $1,378,218 in accrued compensation and benefits as consideration for the issuance of shares of Series H Preferred Stock. During the 12 months preceding October 12, 2021, the Company received an aggregate of $650,550 as consideration for the issuance of convertible notes, and received $288,419 as consideration for the issuance of shares of Common Stock.
The Company, through this Offering and otherwise, is continuing its efforts to raise additional capital in order to be able to pay its liabilities and fund its business activities on a going forward basis, including the pursuit of the Company’s planned research and development activities described in “Use of Proceeds” located elsewhere in this Offering Circular. We provide no assurance that the net proceeds from this Offering will be sufficient for these purposes. The Company regularly evaluates various other measures to satisfy its liquidity needs, including development and other agreements with collaborative partners and, when necessary, seeking to exchange or restructure the Company’s outstanding debt. The Company is evaluating certain changes to its operations and structure to facilitate raising capital from sources that may be interested in financing only discrete aspects of the Company’s development programs. Such changes could include a significant reorganization, which may include the formation of one or more subsidiaries into which one or more programs may be contributed. As a result of the Company’s current financial situation, the Company has limited access to external sources of debt and equity financing. Accordingly, there can be no assurances that the Company will be able to secure additional financing in the amounts necessary to fully fund its operating and debt service requirements. If the Company is unable to access sufficient cash resources, the Company may be forced to discontinue its operations entirely and liquidate.
On October 7, 2021, the Company and Dariusz Nasiek and Sara Nasiek JTTEN (“Nasiek”) entered into a Securities Purchase Agreement pursuant to which Nasiek provided a sum of $103,500 to the Company, in return for a convertible promissory note (the “Nasiek Note”) with a face amount of $115,000, and a common stock purchase warrant exercisable for five years at exercise prices of $0.02 per share on a cash or cashless basis, to purchase up to 5,750,000 shares of the Company’s Common Stock. The Nasiek Note obligates the Company to pay by October 7, 2022, principal amount of $115,000, together with interest at a rate equal to 10% per annum. For more information, see the Company’s Current Report on Form 8-K filed on October 12, 2021, and the exhibits filed therewith.
On November 8, 2021, one convertible note holder converted $80,000 of its convertible note into 4,000,000 shares of Common Stock leaving $32,000 of principal amount and accrued interest unconverted. The Company did not receive any cash proceeds as a result of the conversion.
On November 8, 2021 and November 22, 2021, one warrant holder exercised on a cashless exercise basis, warrants that would have been exercisable into 1,534,042 and 4,300,000 shares of Common Stock, respectively, if exercised on a cash basis, which resulted in the issuance of 511,347 and 1,433,333 shares of Common Stock respectively and resulted in the cancellation of that portion of each warrant exercised (the latter having been exercised in full). The Company did not receive any cash proceeds as a result of the cashless exercises.
Principal Commitments
Employment Agreements
On September 22, 2021, the Board of Directors of the Company approved a second amendment, which became effective on September 27, 2021 (“Amendment No. 2”), of the employment agreement of Timothy Jones, the Company’s President and Chief Executive Officer, and a director of the Company (the “Employment Agreement”). Amendment No. 2 reduces the annual Base Salary from $300,000 to $231,000 from October 1, 2021 through March 31, 2022 and restores the annual Base Salary to $300,000 on April 1, 2022, the payment structure of which is dependent upon the Company raising certain levels of funds. For more information, see the Company’s Current Report on Form 8-K filed on September 28, 2021, and the exhibits filed therewith.
Convertible Notes
On October 7, 2021 and August 31, 2021, the Company and Dariusz Nasiek and Sara Nasiek JTTEN (“Nasiek”) and Barton Asset Management LLC (“Barton”) entered into separate Securities Purchase Agreements pursuant to which Nasiek and Barton each provided a sum of $103,500 to the Company, in return for convertible promissory notes (the “Nasiek and Barton Notes”) each with face amounts of $115,000, and common stock purchase warrants exercisable for five years at exercise prices of $0.02 per share on a cash or cashless basis, to purchase up to 11,500,000 shares of the Company’s Common Stock. The Nasiek and Barton Notes obligate the Company to pay by October 7, 2022 and August 31, 2022, respectively, principal amounts of $115,000 each, together with interest at a rate equal to 10% per annum. For more information, see the Company’s Current Reports on Form 8-K filed on October 12, 2021 and September 3, 2021 respectively, and the exhibits filed therewith.
Payment Settlement Agreement with DNA Healthlink, Inc.
On September 14, 2021, the Company and DNA Healthlink, Inc. (“DNA Healthlink”) entered into a settlement agreement regarding $410,000 in unpaid accounts payable owed by the Company to DNA Healthlink, pursuant to which the amount owed will be paid as follows: twelve monthly payments of $8,000 each commencing on November 15, 2021, followed by twelve monthly payments of $10,000 each commencing on November 15, 2022, followed by twelve monthly payments of $15,000 each commencing on November 15, 2023, followed by one final payment of $14,000 on November 15, 2024, subject to certain conditions. For more information, see the Company’s Current Report on Form 8-K filed on September 20, 2021, and the exhibits filed therewith.
Waivers of Certain Obligations with Respect to this Offering.
Between September 22, 2021 and September 24, 2021, the Company received waivers such that the Company is not required to include in this Offering as selling stockholders, five holders of an aggregate six notes convertible into Common Stock. In addition, a holder of a convertible note renewed its waiver of the Company’s obligation to file a Form 1-A with sixty days of the effective date of the convertible note, which was dated February 17, 2021.
Directors, Executive Officers & Corporate Governance
See Part III. Item 10. Directors, Executive Officers and Corporate Governance in our 2020 Form 10-K.
See Part III. Item 11. Executive Compensation of our 2020 Form 10-K.
| 37 |
Certain Relationships and Related Party Transactions
See Part III. Item 13. Certain Relationships and Related Transactions, and Director Independence in our 2020 Form 10-K.
Security Ownership of Management & Certain Security Holders
The following table sets forth certain information regarding the beneficial ownership of Common Stock as of November 26, 2021, by (i) each person known by the Company to be the beneficial owner of more than 5% of the outstanding Common Stock, (ii) each of the Company’s directors, (iii) each of the Company’s named executive officers, and (iv) all of the Company’s executive officers and directors as a group. Except as indicated in the footnotes to this table, the Company believes that the persons named in this table have sole voting and investment power with respect to the shares of Common Stock indicated. In computing the number and percentage ownership of shares beneficially owned by a person, shares of Common Stock that a person has a right to acquire within sixty (60) days of November 26, 2021 pursuant to options, warrants or other rights are considered as outstanding, while these shares are not considered as outstanding for computing the percentage ownership of any other person or group. The total number of shares of Common Stock outstanding as of November 26, 2021 was 96,341,276.
| Shares Beneficially Owned | ||||||||
| Directors, Officers and 5% Stockholders(a) | Amount and Nature of Beneficial Ownership | Percent of Class | ||||||
| Arnold Lippa Family Trust of 2007 | 22,526,157 | (b) | 20.97 | % | ||||
| Jeff Margolis Trusts | 20,902,174 | (c) | 19.61 | % | ||||
| Directors and Officers: | ||||||||
| Jeff E. Margolis | 20,902,659 | (c) | 19.61 | % | ||||
| Arnold S. Lippa, Ph.D. | 140 | (d) | 0.00 | % | ||||
| Timothy Jones | 2,581,812 | (e) | 2.62 | % | ||||
| Kathryn MacFarlane | 1,264,040 | (f) | 1.30 | % | ||||
| Richard Purcell | 526,306 | (g) | 0.54 | % | ||||
| David Dickason | 200,000 | (h) | 0.21 | % | ||||
| Marc Radin | 5,256,455 | (i) | 5.28 | % | ||||
| All directors and current executive officers as a group (6 persons) | 25,475,191 | 23.01 | % | |||||
| (a) | Except as otherwise indicated, each individual or entity has, or is entitled to have within 60 days of November 26, 2021, sole voting or dispositive power with respect to the shares reported as beneficially owned. |
| (b) | Dr. Lippa is neither the trustee nor the beneficiary of the Arnold Lippa Family Trust of 2007 (the “Lippa Trust”). Morgen Krisch, Dr. Lippa’s daughter, and her two sons are beneficiaries of the Lippa Trust. These holdings include 11,461,716 shares of Common Stock, options to purchase 81,034 shares of Common Stock, warrants exercisable into 10,983,407 shares of Common Stock and 800 shares directly owned by Aurora Capital LLC, which entity is indirectly owned in part by the Lippa Trust and through which the Lippa Trust shares voting and dispositive power over the shares with Jeff E. Margolis. The address of the Lippa Trust is c/o RespireRx Pharmaceuticals Inc., 126 Valley Road, Suite C, Glen Rock, New Jersey 07452. |
| (c) | Mr. Margolis’ holdings are directly held by six trusts, three of which Mr. Margolis is the trustee and the balance of which Mr. Margolis’ spouse is the trustee (the “Margolis Trusts”). These holdings include 10,645,193 shares of Common Stock, options to purchase 66,443 shares of Common Stock, warrants exercisable into 10,190,538 shares of Common Stock and a warrant exercisable into 800 shares of Common Stock directly owned by Aurora Capital LLC, which entity is indirectly owned in part by Mr. Margolis and through which he shares voting and dispositive power over the shares with the Lippa Trust. The address of the Margolis Trusts is c/o RespireRx Pharmaceuticals Inc., 126 Valley Road, Suite C, Glen Rock, New Jersey 07452. |
| 38 |
| (d) | Dr. Lippa’s holdings include 59 shares of Common Stock. |
| (e) | Mr. Jones’ holdings include 440,906 shares of Common Stock, warrants exercisable into 440,906 shares of Common Stock and options to purchase 1,700,000 shares of Common Stock. |
| (f) | Dr. MacFarlane’s holdings include 615 shares of Common Stock and options to purchase 1,263,425 shares of Common Stock. |
| (g) | Mr. Purcell’s holdings include 615 shares of Common Stock and options to purchase 525,691 shares of Common Stock. |
| (h) | Mr. Dickason’s holdings include options to purchase 200,000 shares of Common Stock. |
| (i) | Mr. Radin’s holdings include 2,120,631 shares of Common Stock, warrants to purchase 2,119,679 shares of Common Stock and options to purchase 1,012,387 shares of Common Stock. Mr. Radin’s holdings do not include any shares that may be owned in an individual retirement account held by his spouse. Mr. Radin is not a director or an executive officer of the Company. |
The Company is not aware of any arrangements that may at a subsequent date result in a change of control of the Company.
The following is a general description of our Common Stock and does not purport to be complete. For a complete description of the terms and provisions of our Common Stock, refer to the Company’s Second Restated Certificate of Incorporation, as amended to date (the “Certificate of Incorporation”) and By-Laws of the Company, as amended (the “Bylaws”), each of which is an exhibit incorporated by reference into the Offering Statement of which this Offering Circular is a part. This summary is qualified in its entirety by reference to these documents.
Authorized and Outstanding Capital Stock
The Company is authorized to issue a total of 2,005,000,000 shares of capital stock, with a par value of $0.001 per share. Of the authorized amount, 2,000,000,000 of the shares are designated as Common Stock and 5,000,000 of the shares are designated as preferred stock.
As of September 30, 2021, there were 90,396,596 shares of Common Stock issued and outstanding.
Description of Common Stock
General. Each share of the Company’s Common Stock has the same rights and privileges. Holders of the Common Stock do not have any preferences or any preemptive, redemption, subscription, conversion or exchange rights. All outstanding shares of Common Stock are fully paid and non-assessable. The Company’s Common Stock is quoted on the OTCQB, under the symbol “RSPI.”
Voting Rights. The holders of Common Stock are entitled to vote upon all matters submitted to a vote of stockholders and are entitled to one vote for each share of Common Stock held. There is no cumulative voting.
Dividends. The Company has never paid cash dividends on its Common Stock and does not anticipate paying such dividends in the foreseeable future. The payment of dividends, if any, will be determined by the Board of Directors in light of conditions then existing and may be paid on the Common Stock subject to the prior rights and preferences, if any, applicable to shares of preferred stock or any series of preferred stock, when and if declared by the Board of Directors, out of funds legally available therefor.
Liquidation and Distribution. If the Company voluntarily or involuntarily liquidates, dissolves or winds-up, or upon any distribution of assets, the holders of Common Stock will be entitled to receive, after distribution in full of the preferential amounts, if any, to be distributed to the holders of preferred stock or any series of preferred stock, all of the remaining assets available for distribution equally and ratably in proportion to the number of shares of Common Stock held by them.
Material Limitation or Qualification of Rights of Common Stock
Preferred Stock, Generally. The Company may issue preferred stock with such powers, preferences, rights, qualifications, limitations, and restrictions as the Board of Directors may, without prior stockholder approval, establish. The existence, and potential future issuance, of shares of preferred stock by the Company could result in substantial dilution of the economic and governance rights of holders of the Company’s common stock.
| 39 |
As of September 30, 2021, the Company’s authorized shares of preferred stock are designated into series as follows: 3,000 shares of Series H 2% Voting, Non-Participating Convertible Preferred Stock, 37,500 shares as Series B Convertible Preferred Stock (“Series B Preferred Stock”), 1,700 shares as Series G 1.5% Convertible Preferred Stock (“Series G Preferred Stock”), 1,250,000 shares as 9% Cumulative Convertible Preferred Stock (“9% Preferred Stock”), 205,000 shares as Series A Junior Participating Preferred Stock (“Series A Preferred Stock”), and 3,505,800 shares are undesignated and may be issued with such rights and powers as the Board of Directors may designate.
Series H Preferred Stock. As of September 30, 2021, there were no shares of Series H Preferred Stock are issued and outstanding or accrued as dividends as all outstanding shares of Series H Preferred Stock inclusive of accrued dividends converted into units that resulted in the issuance of 25,377,426 shares of Common Stock and warrants to purchase 25,377,426 shares of Common Stock. Each share of Series H Preferred Stock is convertible into 15,625 units at an effective conversion price of $0.064 per unit, with each unit comprising one share of Common Stock and one warrant exercisable for one share of Common Stock. Each share of Series H Preferred Stock entitles the holder to that number of votes equal to two times the number of shares of Common Stock into which it is convertible. In the event of any liquidation or winding up of the Company prior to and in preference to any junior securities, the holders of the Series H Preferred Stock will be entitled to receive in preference to the holders of any junior securities a per share amount equal to the $0.001, plus any accrued and unpaid dividends.
Series B Preferred Stock. As of September 30, 2021, 37,500 shares of Series B Preferred Stock are issued and outstanding. Each share of Series B Preferred Stock is convertible into approximately 0.000030 shares of common stock at an effective conversion price of $22,083.75 per share of common stock, which is subject to adjustment under certain circumstances. As of September 30, 2021, the shares of Series B Preferred Stock outstanding are convertible into 1 share of Common Stock. Shares of Series B Preferred Stock do not entitle the holder to voting rights. The Company may redeem the Series B Preferred Stock for $25,001, equivalent to $0.6667 per share, an amount equal to the liquidation preference, at any time upon 30 days prior notice.
Series G Preferred Stock. As of September 30, 2021, no shares of Series G Preferred Stock are issued and outstanding. If issued, each share of Series G Preferred Stock is convertible into that number of shares of Common Stock determined by dividing $1,000 by an initial conversion price of $0.033. The conversion price with respect to a share of Series G Preferred Stock is subject to adjustment upon certain events that occur while such share is outstanding, pursuant to Section 7 of the Certificate of Designation, Preferences, Rights and Limitations of Series G 1.5% Convertible Preferred Stock (incorporated by reference to Exhibit 3.1 to the Company’s Current Report on form 8-K filed on March 24, 2014). As of December 31, 2020, the conversion price with respect to Series G Preferred Stock is not subject to adjustment because no shares of Series G Preferred Stock are outstanding. If issued, each outstanding share of Series G Preferred Stock, prior to the date such share is eligible for conversion, entitles the holder to 30,303 votes per share (which may be subject to adjustment as described above), and thereafter, each share entitles the holder to voting rights on an as-converted basis.
9% Preferred Stock. As of September 30, 2021, no shares of 9% Preferred Stock are issued and outstanding. If issued, each share of 9% Preferred Stock is convertible into shares of common stock according to a conversion rate subject to adjustment upon the occurrence of certain events, including a reverse stock split, as set forth under our Certificate of Incorporation. Thereunder, each share of 9% Preferred Stock is convertible into that number of shares of common stock determined by dividing $1.00 by a conversion rate of $1.50, subject to adjustment pursuant to the reverse stock splits effected by the Company on September 1, 2016 and January 5, 2021, whereby, on September 1, 2016 each 325 shares of Common Stock was exchanged and combined into one share of Common Stock and on January 5, 2021, each 10 shares of Common Stock was exchanged and combined into one share of Common Stock. Shares of 9% Preferred Stock do not entitle the holder to voting rights.
Series A Preferred Stock. As of September 30, 2021, no shares of Series A Preferred Stock are issued and outstanding. Shares of Series A Preferred Stock do not entitle the holder to voting rights, except to the extent the holder would be entitled to vote with the holders of Common Stock as set forth in the Certificate of Designation for the Series A Preferred Stock (see our Certificate of Incorporation).
Anti-Takeover Provisions in the Certificate of Incorporation and Bylaws
Certain provisions of our Certificate of Incorporation and Bylaws summarized below may delay, defer or prevent a tender offer or takeover attempt, including attempts that might result in a premium over the market price for the Company’s securities.
Our Certificate of Incorporation and Bylaws provide: (i) that the Company may issue preferred stock with such powers, preferences, rights, qualifications, limitations, and restrictions as the Board of Directors may, without prior stockholder approval, establish, as described above; and (ii) that special meetings of stockholders may only be called by the chairman of the Board of Directors, the president, the secretary, a majority of the members of the Board of Directors or the holders of a majority of the shares of Common Stock then outstanding.
| 40 |
RespireRx has never and currently does not intend to declare dividends on its Common Stock. Dividends may accrue, may be paid in cash or may be paid in-kind with respect to certain series of preferred stock, none of such series being currently outstanding.
Interests of Named Experts and Counsel
The validity of the issuance of Common Stock qualified hereby is passed on for the Company by its outside counsel, Faegre Drinker Biddle & Reath LLP. In January 2017, the Company issued options to purchase 1,000 shares of its Common Stock under the Amended and Restated RespireRx Pharmaceuticals Inc. 2015 Stock and Stock Option Plan, as amended, to Faegre Drinker Biddle & Reath LLP, as partial payment for services previously provided and invoiced. The options expire on January 17, 2022 and have an exercise price of $39.00 per share.
Information Incorporated by Reference
The SEC allows us to incorporate by reference the information we file with it, which means that we can disclose important information to you by referring you to another document that we have filed separately with the SEC. We hereby incorporate by reference the following information or documents into this Offering Circular:
Any information in any of the foregoing documents will automatically be deemed to be modified or superseded to the extent that information in this Offering Circular or in a later filed document that is incorporated or deemed to be incorporated herein by reference modifies or replaces such information.
We urge you to carefully read this Offering Circular and the documents incorporated by reference herein, before buying any of the Shares being offered under this Offering Circular. This Offering Circular may add or update information contained in the documents incorporated by reference herein. To the extent that any statement that we make in this Offering Circular is inconsistent with statements made in the documents incorporated by reference herein, you should rely on the information in this Offering Circular and the statements made in this Offering Circular will be deemed to modify or supersede those made in the documents incorporated by reference herein.
You should rely only on the information contained in this Offering Circular or incorporated herein by reference. We have not authorized anyone to provide you with different information. No dealer, salesperson or other person is authorized to give any information or to represent anything not contained in this Offering Circular or incorporated herein by reference. You should not rely on any unauthorized information or representation. This Offering Circular is an offer to sell only the Shares offered hereby, and only under circumstances and in jurisdictions where it is lawful to do so. You should assume that the information in this Offering Circular is accurate only as of the date on the front of the applicable document and that any information we have incorporated by reference is accurate only as of the date of the document incorporated by reference, regardless of the time of delivery of this Offering Circular, or any sale of a security.
| 41 |
We further note that the representations, warranties and covenants made by us in any agreement that is filed as an exhibit to any document that is incorporated by reference in this Offering Circular were made solely for the benefit of the parties to such agreement, including, in some cases, for the purpose of allocating risk among the parties to such agreements, and should not be deemed to be a representation, warranty or covenant to you. Moreover, such representations, warranties or covenants were accurate only as of the date when made. Accordingly, such representations, warranties and covenants should not be relied on as accurately representing the current state of our affairs.
As used in this Offering Circular, all references to “RespireRx,” the “Company,” “we,” “our,” “Shares” “capital stock,” “Common Stock,” “Series B Preferred Stock,” “Preferred Stock” or “stockholders,” applies only to RespireRx Pharmaceuticals Inc. As used in this Offering Circular, the terms “consolidated we,” “consolidated our” or words of like import mean RespireRx Pharmaceuticals Inc. and its direct wholly-owned subsidiary, Pier Pharmaceutical, Inc. Notwithstanding the foregoing, references to the company, we, our and similar terms that appear in the consolidated financial statements in our annual reports on Form 10-K and in our condensed consolidated financial statements in our quarterly reports on Form 10-Q, refer to RespireRx Pharmaceuticals Inc. and its direct wholly-owned subsidiary Pier Pharmaceutical, Inc. All references in this Offering Circular to “years” and “fiscal years” means the twelve-month period ended December 31st, unless the context indicates otherwise.
Upon written or oral request, we will provide you without charge a copy of any or all of the documents that are incorporated by reference into this Offering Circular, including but limited to financial statement information and exhibits which are specifically incorporated by reference into such documents. Requests should be directed to: RespireRx Pharmaceuticals Inc., Attention: Jeff Eliot Margolis, 126 Valley Road, Suite C, Glen Rock, NJ 07452, jmargolis@respirerx.com or 917-834-7206. You may access this information at www.respirerx.com and at http://compliance-sec.com/secfilings/company/corx/filings.html.
Where You Can Find More Information
We have filed with the SEC a Regulation A Offering Statement on Form 1-A under the Securities Act of 1993, as amended, with respect to the shares of Common Stock offered hereby. This Offering Circular, which constitutes a part of the Offering Statement, does not contain all of the information set forth in the Offering Statement or the exhibits and schedules filed therewith. For further information about us and the Common Stock offered hereby, we refer you to the Offering Statement and the exhibits and schedules filed therewith. Statements contained in this Offering Circular regarding the contents of any contract or other document that is filed as an exhibit to the Offering Statement are not necessarily complete, and each such statement is qualified in all respects by reference to the full text of such contract or other document filed as an exhibit to the Offering Statement. We are currently required to file periodic reports, proxy statements, and other information with the SEC pursuant to the Securities Exchange Act of 1934. You may read and copy this information at the SEC’s Public Reference Room, 100 F Street, N.E., Room 1580, Washington, D.C. 20549. You may obtain information on the operation of the Public Reference Room by calling the SEC at 1-800-SEC-0330. The SEC also maintains an Internet website that contains reports, proxy statements and other information about issuers, including us, that file electronically with the SEC. The address of this site is www.sec.gov.
| 42 |
Financial Statements and Exhibits
RESPIRERX PHARMACEUTICALS INC.
AND SUBSIDIARY
INDEX TO CONSOLIDATED FINANCIAL STATEMENTS
(INCLUDING REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM)
Audited consolidated financial statements as of and for the years ended December 31, 2020 and 2019
Unaudited interim condensed consolidated financial statements as of September 30, 2021 and for the three-months and nine- months ended September 30, 2021 and 2020.
| F-1 |
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
To the Stockholders and Board of Directors
RespireRx Pharmaceuticals Inc. and Subsidiary
Opinion on the Consolidated Financial Statements
We have audited the accompanying consolidated balance sheets of RespireRx Pharmaceuticals Inc. and Subsidiary (the “Company”) as of December 31, 2020 and 2019, the related consolidated statements of operations, stockholders’ deficiency, and cash flows for each of the years then ended, and the related notes (collectively, the “consolidated financial statements”). In our opinion, the consolidated financial statements present fairly, in all material respects, the consolidated financial position of the Company as of December 31, 2020 and 2019, and the consolidated results of its operations and its cash flows for each of the years then ended, in conformity with generally accepted accounting principles generally accepted in the United States of America.
Going Concern
The accompanying consolidated financial statements have been prepared assuming that the Company will continue as a going concern. As discussed in Note 2 to the consolidated financial statements, the Company has experienced recurring losses, negative cash flows from operations, has limited capital resources, and a net stockholders’ deficiency. These matters raise substantial doubt about the Company’s ability to continue as a going concern. Management’s plans in regard to these matters are also described in Note 2. The consolidated financial statements do not include any adjustments that might result from the outcome of this uncertainty.
Basis for Opinion
These consolidated financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on the Company’s consolidated financial statements based on our audits. We are a public accounting firm registered with the Public Company Accounting Oversight Board (United States) (“PCAOB”) and are required to be independent with respect to the Company in accordance with the U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our audits in accordance with the standards of the PCAOB. Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the consolidated financial statements are free of material misstatement, whether due to error or fraud. The Company is not required to have, nor were we engaged to perform, an audit of its internal control over financial reporting. As part of our audits, we are required to obtain an understanding of internal control over financial reporting but not for the purpose of expressing an opinion on the effectiveness of the Company’s internal control over financial reporting. Accordingly, we express no such opinion.
Our audits included performing procedures to assess the risks of material misstatement of the consolidated financial statements, whether due to error or fraud, and performing procedures that respond to those risks. Such procedures included examining, on a test basis, evidence regarding the amounts and disclosures in the consolidated financial statements. Our audits also included evaluating the accounting principles used and significant estimates made by management, as well as evaluating the overall presentation of the consolidated financial statements. We believe that our audits provide a reasonable basis for our opinion.
| F-2 |
Critical Audit Matters
The critical audit matters communicated below are matters arising from the current period audit of the consolidated financial statements that were communicated or required to be communicated to the audit committee and that: (1) relate to accounts or disclosures that are material to the consolidated financial statements and (2) involved our especially challenging, subjective, or complex judgments. The communication of the critical audit matters does not alter in any way our opinion on the consolidated financial statements, taken as a whole, and we are not, by communicating the critical audit matters below, providing separate opinions on the critical audit matters or on the accounts or disclosures to which they relate.
Share Based Compensation
Critical Audit Matter Description
The Company issues stock options to employees and vendors. Management uses the Black-Scholes option-pricing model to estimate the fair value of its stock options. The Black-Scholes option-pricing model involves the use of significant estimates, including the following:
| ■ | Risk-free interest rate; |
| ■ | Expected share price volatility; |
| ■ | Expected dividend yield; and |
| ■ | Expected life of the award. |
Given the significant estimates involved in determining the fair value of stock options, the related audit effort in evaluating management’s estimates for the inputs to the Black-Scholes pricing model was extensive and required a high degree of auditor judgment.
How the Critical Audit Matter was Addressed in the Audit
We obtained an understanding of the management’s process to estimate the fair value of stock options, including how management develops each of the estimates required as inputs to the Black-Scholes option-pricing model. We applied the following audit procedures related to testing management’s estimates utilized in the Black-Scholes option-pricing model:
| ■ | We compared the Company’s risk-free interest rate used to the comparable United States Treasury yield for a term comparable to option’s estimated life. | |
| ■ | We recalculated the Company’s historical share price volatility for a term of 12 months prior to the grant date because management considered the volatility for this period to be a better reflection of future value than the historical share price volatility of the term of the options. | |
| ■ | We performed a look-back of the Company’s previously issued dividends, noting there were none. We inquired with management of the Company who informed us that no future dividends were currently anticipated. | |
| ■ | We agreed the inputs used for the life of the award to management’s estimate of the expected life, which is the contractual life as no options have been exercised recently. |
| F-3 |
Accounting for Complex Debt Transactions
Critical Audit Matter Description
During the year ended December 31, 2020, the Company entered into several convertible notes payable that included original issue discounts, beneficial conversion features, and warrants. The proceeds of the convertible notes payable are allocated to the components of the convertible debt instrument in accordance with ASC 470-20, Debt with Conversion and Other Options. Management used the Black-Scholes option-pricing model to estimate the fair value of the warrants issued with the convertible notes and allocated the proceeds to the warrants and the debt host based on a relative fair value basis. The Black-Scholes option-pricing model involves the use of significant estimates, including the following:
| ● | Risk-free interest rate; | |
| ● | Expected share price volatility; | |
| ● | Expected dividend yield; and | |
| ● | Contractual life of the award. |
Given the significant estimates involved in determining the individual components of the debt instrument and the related debt discounts resulting from the relative fair value calculation for the warrants and intrinsic value of the beneficial conversion features, the related audit effort in evaluating management’s estimates in determining those items was extensive and required a high degree of auditor judgment.
How the Critical Audit Matter was Addressed in the Audit
We obtained an understanding over management’s process to determine the individual components of the debt instrument and the methodology to calculate the relative fair value of the warrants and beneficial conversion features, in accordance with the applicable accounting standards. This also included assessing how management develops each of the estimates for the inputs to the Black-Scholes option-pricing model. We applied the following audit procedures related to testing the management’s estimates utilized in the Black-Scholes option-pricing model for valuing the warrants:
| ■ | We compared the Company’s risk-free interest rate used to the comparable United States Treasury yield for a term comparable to the warrants’ remaining contractual term. | |
| ■ | We recalculated the Company’s historical share price volatility for a term of 12 months prior to the grant date because management considered the volatility for this period to be a better reflection of future value than the historical share price volatility of the term of the options. | |
| ■ | We performed a look-back of the Company’s previously issued dividends, noting there were none. We inquired with management of the Company who informed us that no future dividends were currently anticipated. | |
| ■ | We agreed the inputs used for the term of the warrants to the contractual term of the warrant. |
We also reviewed management’s relative fair value calculation used to determine the components of the debt instrument and the values assigned to each as follows:
| ■ | We obtained a copy of the convertible debt agreement to understand its terms, noting management properly identified the components of the debt instrument. | |
| ■ | We agreed the proceeds of the notes to confirmation letters with the note holder or to deposits in the banking records. | |
| ■ | We evaluated the accounting methodology to assign values to the individual debt components determining it was consistent with the applicable accounting standards. | |
| ■ | We agreed the fair value assigned to the warrants to the fair value calculated using the Black-Scholes option-pricing model. | |
| ■ | We recalculated the relative fair value assigned to the warrants and host debt instrument. | |
| ■ | We agreed the conversion price to the convertible note agreement and recalculated the intrinsic value of the beneficial conversion features, noting it was properly recorded as a debt discount. |
| HASKELL & WHITE LLP |
We have served as the Company’s auditor since 2004.
Irvine, California
April 15, 2021
| F-4 |
RESPIRERX PHARMACEUTICALS INC.
AND SUBSIDIARY
| December 31, | ||||||||
| 2020 | 2019 | |||||||
| ASSETS | ||||||||
| Current assets: | ||||||||
| Cash and cash equivalents | $ | 825 | $ | 16,690 | ||||
| Deferred financing costs | 52,609 | - | ||||||
| Prepaid expenses, including current portion of long-term prepaid insurance of $0 at December 31, 2020 and $10,586 at December 31, 2019 | 31,653 | 28,638 | ||||||
| Total current assets | 85,087 | 45,328 | ||||||
| Total assets | $ | 85,087 | $ | 45,328 | ||||
| LIABILITIES AND STOCKHOLDERS’ DEFICIENCY | ||||||||
| Current liabilities: | ||||||||
| Accounts payable and accrued expenses, including $635,146 and $476,671 payable to related parties at December 31, 2020 and 2019, respectively | $ | 4,923,947 | $ | 3,772,030 | ||||
| Accrued compensation and related expenses | 1,540,809 | 2,083,841 | ||||||
Convertible notes payable, currently due and payable on demand, including accrued interest of $85,693 and $113,304 at December 31, 2020 and 2019, respectively, (of which $48,700, including accrued interest of $23,700, was deemed to be in default at December 31, 2020) (Note 4) | 414,860 | 551,591 | ||||||
| Note payable to SY Corporation, including accrued interest of $411,385 and $363,280 at December 31, 2020 and 2019, respectively (payment obligation currently in default – Note 4) | 864,551 | 766,236 | ||||||
| Notes and advances payable to officers, including accrued interest of $46,717 and $35,388 at December 31, 2020 and 2019, respectively (Note 4) | 213,067 | 142,238 | ||||||
| Notes payable to former officer, including accrued interest of $58,965 and $41,977 as of December 31, 2020 and December 31, 2019, respectively (Note 4) | 186,565 | 169,577 | ||||||
| Other short-term notes payable | 4,608 | 4,634 | ||||||
| Total current liabilities | 8,148,407 | 7,490,147 | ||||||
| Commitments and contingencies (Note 9) | ||||||||
| Stockholders’ deficiency: (Note 6) | ||||||||
| Series B convertible preferred stock, $0.001 par value; $0.6667 per share liquidation preference; aggregate liquidation preference $25,001; shares authorized: 37,500; shares issued and outstanding: 37,500; common shares issuable upon conversion at 0.000030 common shares per Series B share: 1 | 21,703 | 21,703 | ||||||
| Common stock, $0.001 par value; shares authorized: 2,000,000,000; shares issued and outstanding: 71,271,095 and 417,507 at December 31, 2020 and 2019, respectively (reflected on a post 10 for 1 reverse stock split basis which occurred on January 5, 2021) | 71,271 | 418 | ||||||
| Additional paid-in capital | 162,654,002 | 159,042,145 | ||||||
| Accumulated deficit | (170,810,296 | ) | (166,509,085 | ) | ||||
| Total stockholders’ deficiency | (8,063,320 | ) | (7,444,819 | ) | ||||
| Total liabilities and stockholders’ deficiency | $ | 85,087 | $ | 45,328 | ||||
See accompanying notes to consolidated financial statements and
report of independent registered public accounting firm.
| F-5 |
RESPIRERX PHARMACEUTICALS INC.
AND SUBSIDIARY
CONSOLIDATED STATEMENTS OF OPERATIONS
| Years Ended December 31, | ||||||||
| 2020 | 2019 | |||||||
| Operating expenses: | ||||||||
| General and administrative, including $1,230,370 and $485,332 to related parties for the years ended December 31, 2020 and 2019, respectively | $ | 2,676,860 | $ | 1,137,175 | ||||
| Research and development, including $490,850 and $490,908 to related parties for the years ended December 31, 2020 and 2019, respectively | 638,275 | 599,329 | ||||||
| Total operating costs and expenses | 3,315,135 | 1,736,504 | ||||||
| Loss from operations | (3,315,135 | ) | (1,736,504 | ) | ||||
| Loss on extinguishment of debt and other liabilities in exchange for equity | (389,902 | ) |
- | |||||
| Interest expense, including $11,329 and $60,135 to related parties for the years ended December 31, 2020 and 2019, respectively | (545,675 | ) | (404,661 | ) | ||||
| Foreign currency transaction (loss) gain | (50,499 | ) | 26,132 | |||||
| Net loss | $ | (4,301,211 | ) | $ | (2,115,033 | ) | ||
| Deemed dividends from warrant anti-dilution provisions | $ | (1,440,214 | ) | $ | - | |||
Net loss attributable to common shareholders | $ | (5,741,425 | ) | $ | (2,115,033 | ) | ||
| Net loss per common share - basic and diluted respectively (reflected on a post 10 for 1 reverse stock split basis which occurred on January 5, 2021) | $ | (0.22) | $ | (5.41 | ) | |||
| Weighted average common shares outstanding - basic and diluted respectively (reflected on a post 10 for 1 reverse stock split basis which occurred on January 5, 2021) | 25,855,664 | 390,848 | ||||||
See accompanying notes to consolidated financial statements and
report of independent registered public accounting firm.
| F-6 |
RESPIRERX PHARMACEUTICALS INC.
AND SUBSIDIARY
CONSOLIDATED STATEMENTS OF STOCKHOLDERS’ DEFICIENCY
Years Ended December 31, 2020 and 2019
| Series B and Series H | ||||||||||||||||||||||||||||
| Convertible | ||||||||||||||||||||||||||||
| Preferred Stock | Common Stock | Additional | Total | |||||||||||||||||||||||||
| Shares | Amount | Shares | Par Value | Paid-in
Capital | Accumulated
Deficit | Stockholders’
Deficiency | ||||||||||||||||||||||
| Balance at December 31, 2018 | 37,500 | $ | 21,703 | 387,207 | $ | 387 | $ | 158,638,707 | $ | (164,394,052 | ) | $ | (5,733,255 | ) | ||||||||||||||
| Warrants issued with respect to convertible notes issued from January through March 2019 | 45,812 | 45,812 | ||||||||||||||||||||||||||
| Common stock issued related to convertible notes | 1,750 | 2 | 3,331 | 3,333 | ||||||||||||||||||||||||
| Discounts associated with convertible note issuances from April through November 2019 | 329,019 | 329,019 | ||||||||||||||||||||||||||
| Common stock issued as partial settlement of convertible notes issued from April through May 2019 | 28,550 | 29 | 25,276 | 25,305 | ||||||||||||||||||||||||
| Net Loss | $ | (2,115,033 | ) | $ | (2,115,033 | ) | ||||||||||||||||||||||
| Balance at December 31, 2019 | 37,500 | $ | 21,703 | 417,507 | $ | 418 | $ | 159,042,145 | $ | (166,509,085 | ) | $ | (7,444,819 | ) | ||||||||||||||
| Issuance of Common Stock for payment of accrued compensation | 900,000 | 900 | 305,100 | 306,000 | ||||||||||||||||||||||||
| Issuances of Series H Preferred Stock payment of accrued compensation | 1,383 | $ | 1 | 1,378,217 | 1,378,218 | |||||||||||||||||||||||
| Issuance of Series H Preferred Stock for payment of accounts payable | 241 | $ | 0 | 307,015 | 307,015 | |||||||||||||||||||||||
| Conversion of Series H Preferred Stock to Common Stock | (1,624 | ) | $ | (1 | ) | (1,685,232 | ) | (1,685,233 | ) | |||||||||||||||||||
| Issuance of Common Stock and Warrants for Conversion of Series H Preferred Stock | 25,377,426 | $ | 25,377 | 1,659,856 | 1,685,233 | |||||||||||||||||||||||
| Sale of Common Stock, net of costs | 7,900,000 | $ | 7,900 | 78,237 | 86,137 | |||||||||||||||||||||||
| Note payable issued with Common Stock | (40,000 | ) | (40,000 | ) | ||||||||||||||||||||||||
| Note discounts | 90,000 | 90,000 | ||||||||||||||||||||||||||
| Note payable conversions | 26,291,373 | $ | 26,291 | 1,100,347 | 1,126,638 | |||||||||||||||||||||||
| Option grants | 384,250 | 384,250 | ||||||||||||||||||||||||||
| Cashless Warrant exercises | 10,384,789 | $ | 10,385 | (10,385 | ) | - | ||||||||||||||||||||||
| Warrants issued with convertible debt | 44,452 | 44.452 | ||||||||||||||||||||||||||
| Net Loss | $ | (4,301,211 | ) | $ | (4,301,211 | ) | ||||||||||||||||||||||
| Balance at December 31, 2020 | 37,500 | $ | 21,703 | 71,271,095 | $ | 71,271 | $ | 162,654,002 | $ | (170,810,296 | ) | $ | (8,063,320 | ) | ||||||||||||||
See accompanying notes to consolidated financial statements and
report of independent registered public accounting firm.
| F-7 |
RESPIRERX PHARMACEUTICALS INC.
AND SUBSIDIARY
CONSOLIDATED STATEMENTS OF CASH FLOWS
| Years Ended December 31, | ||||||||
| 2020 | 2019 | |||||||
| Cash flows from operating activities: | ||||||||
| Net loss | $ | (4,301,211 | ) | $ | (2,115,033 | ) | ||
| Adjustments to reconcile net loss to net cash used in operating activities: | ||||||||
| Amortization of debt discounts related to convertible notes payable | 374,080 | 215,575 | ||||||
| Costs associated with convertible note conversion paid with common stock | 49,219 | 750 | ||||||
| Loss on extinguishment of debt | 323,996 | - | ||||||
| Loss on extinguishment of other liabilities | 65,906 | - | ||||||
| Stock-based compensation and fees included in - | ||||||||
| General and administrative expenses | 345,500 | - | ||||||
| Research and development expenses and vesting options | 38,750 | - | ||||||
| Foreign currency transaction loss (gain) | 50,211 | (26,132 | ) | |||||
| Changes in operating assets and liabilities: | ||||||||
| (Increase) decrease in - | ||||||||
| Prepaid expenses and advanced clinical research payments | (3,014 | ) | 13,355 | |||||
| Increase (decrease) in - | ||||||||
| Accounts payable and accrued expenses | 1,260,922 | 524,324 | ||||||
| Accrued compensation and related expenses | 1,141,186 | 779,407 | ||||||
| Accrued interest payable | 141,454 | 120,009 | ||||||
| Net cash used in operating activities | (513,001 | ) | (487,745 | ) | ||||
| Cash flows from financing activities: | ||||||||
| Proceeds from sale of common stock | 162,886 | - | ||||||
| Proceeds from officer notes | 59,500 | 22,751 | ||||||
| Proceeds from issuance of notes payable | 274,750 | 478,150 | ||||||
| Net cash provided by financing activities | 497,136 | 471,151 | ||||||
| Cash and cash equivalents: | ||||||||
| Net decrease | (15,865 | ) | (16,594 | ) | ||||
| Balance at beginning of period | 16,690 | 33,284 | ||||||
| Balance at end of period | $ | 825 | $ | 16,690 | ||||
(Continued)
| F-8 |
RESPIRERX PHARMACEUTICALS INC.
AND SUBSIDIARY
CONSOLIDATED STATEMENTS OF CASH FLOWS
(Continued)
| Years Ended December 31, | ||||||||
| 2020 | 2019 | |||||||
| Supplemental disclosures of cash flow information: | ||||||||
| Cash paid for - | ||||||||
| Interest | $ | 6,466 | $ | 5,130 | ||||
| Non-cash financing activities: | ||||||||
| Issuance of common stock in exchange for extinguishment of Convertible Notes Payable | $ | 694,946 | $ | - | ||||
| Conversion fees paid with common stock upon principal payment on convertible notes payable | $ | 30,632 | $ | 750 | ||||
| Accounts payable and accrued expenses extinguished with common stock options | $ | 241,109 | $ | - | ||||
| Issuance of common stock in payment of accrued compensation | $ | 1,684,218 | $ | - | ||||
| Issuance of commitment note for equity line | $ | 40,000 | $ | - | ||||
| Issuance of warrants with convertible notes | $ | 44,451 | $ | - | ||||
| Beneficial conversion feature associated with convertible notes | $ | 90,000 | $ | - | ||||
| Short-term note payable issued in connection with financing of directors and officers insurance policy | $ | 70,762 | $ | 61,746 | ||||
| Short-term note payable issued in connection with financing of clinical trial and other office insurance policies | $ | 9,215 | $ | 9,322 | ||||
| Interest liability paid with common stock | 11,760 | - | ||||||
See accompanying notes to consolidated financial statements and
report of independent registered public accounting firm.
| F-9 |
RESPIRERX PHARMACEUTICALS INC.
AND SUBSIDIARY
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
Years Ended December 31, 2020 and 2019
1. Organization and Basis of Presentation
Organization
RespireRx Pharmaceuticals Inc. (“RespireRx”) was formed in 1987 under the name Cortex Pharmaceuticals, Inc. to engage in the discovery, development and commercialization of innovative pharmaceuticals for the treatment of neurological and psychiatric disorders. On December 16, 2015, RespireRx filed a Certificate of Amendment to its Second Restated Certificate of Incorporation (as amended, the “Certificate of Incorporation”) with the Secretary of State of the State of Delaware to amend its Second Restated Certificate of Incorporation to change its name from Cortex Pharmaceuticals, Inc. to RespireRx Pharmaceuticals Inc. In August 2012, RespireRx acquired Pier Pharmaceuticals, Inc. (“Pier”), which is now a wholly owned subsidiary. Pier was a clinical stage biopharmaceutical company developing a pharmacologic treatment for obstructive sleep apnea (“OSA”) and had been engaged in research and clinical development activities which activities are now in RespireRx.
Basis of Presentation
The consolidated financial statements are of RespireRx and its wholly owned subsidiary, Pier.
2. Business
The mission of the Company is to develop innovative and revolutionary treatments to combat disorders caused by disruption of neuronal signaling. We are developing treatment options that address conditions that affect millions of people, but for which there are limited or poor treatment options, including obstructive sleep apnea (“OSA”), attention deficit hyperactivity disorder (“ADHD”) epilepsy, chronic pain, including inflammatory and neuropathic pain, recovery from spinal cord injury (“SCI”), as well as other areas of interest based on results of preclinical and clinical studies to date.
RespireRx is developing a pipeline of new drug products based on our broad patent portfolios across two distinct drug platforms:
| (i) | our pharmaceutical cannabinoids platform (which we refer to as ResolutionRx), including dronabinol, which acts upon the nervous system’s endogenous cannabinoid receptors, and | |
| (ii) | our neuromodulators platform (which we refer to as EndeavourRx) is made up of two programs: (a) our ampakines program, which is developing proprietary compounds that are positive allosteric modulators (“PAMs”) of AMPA-type glutamate receptors to promote neuronal function and (b) our GABAkines program, which is developing proprietary compounds that are PAMs of GABAA receptors, and which was recently established pursuant to our entry with the University of Wisconsin-Milwaukee Research Foundation, Inc., an affiliate of the University of Wisconsin-Milwaukee (“UWMRF”), into a patent license agreement (the “UWMRF Patent License Agreement”). |
| F-10 |
Financing our Platforms
Our major challenge has been to raise substantial equity or equity-linked financing to support research and development plans for our cannabinoid and neuromodulator platforms, while minimizing the dilutive effect to pre-existing stockholders. At present, we believe that we are hindered primarily by our public corporate structure, our OTCQB listing, and low market capitalization as a result of our low stock price.
For this reason, the Company has effected an internal restructuring plan through which our two drug platforms have been reorganized into separate business units and may in the future, be organized into subsidiaries of RespireRx. We believe that by creating one or more subsidiaries to further the aims of ResolutionRx and EndeavourRx, it may be possible, through separate finance channels, to optimize the asset values of each. We are also planning to commence a securities offering by the Company pursuant to Regulation A by filing a Form 1-A.
Going Concern
The Company’s consolidated financial statements have been presented on the basis that it is a going concern, which contemplates the realization of assets and satisfaction of liabilities in the normal course of business. The Company has incurred net losses of $4,301,211 and $2,115,033 for the fiscal years ended December 31, 2020 and 2019, respectively, and negative operating cash flows of $513,001 and $487,745 for the fiscal years ended December 31, 2020 and 2019, respectively. The Company also had a stockholders’ deficiency of $8,063,320 at December 31, 2020 and expects to continue to incur net losses and negative operating cash flows for at least the next few years. As a result, management has concluded that there is substantial doubt about the Company’s ability to continue as a going concern, and the Company’s independent registered public accounting firm, in its report on the Company’s consolidated financial statements for the year ended December 31, 2020, expressed substantial doubt about the Company’s ability to continue as a going concern.
The Company is currently, and has for some time, been in significant financial distress. It has extremely limited cash resources and current assets and has no ongoing source of sustainable revenue. Management is continuing to address various aspects of the Company’s operations and obligations, including, without limitation, debt obligations, financing requirements, intellectual property, licensing agreements, legal and patent matters and regulatory compliance, and has taken steps to continue to raise new debt and equity capital to fund the Company’s business activities from both related and unrelated parties.
| F-11 |
The Company is continuing its efforts to raise additional capital in order to be able to pay its liabilities and fund its business activities on a going forward basis, including the pursuit of the Company’s planned research and development activities. The Company regularly evaluates various measures to satisfy the Company’s liquidity needs, including development and other agreements with collaborative partners and, when necessary, seeking to exchange or restructure the Company’s outstanding securities. The Company is evaluating certain changes to its operations and structure to facilitate raising capital from sources that may be interested in financing only discrete aspects of the Company’s development programs. Such changes could include a significant reorganization, which may include the formation of one or more subsidiaries into which one or more programs may be contributed. As a result of the Company’s current financial situation, the Company has limited access to external sources of debt and equity financing. Accordingly, there can be no assurances that the Company will be able to secure additional financing in the amounts necessary to fully fund its operating and debt service requirements. If the Company is unable to access sufficient cash resources, the Company may be forced to discontinue its operations entirely and liquidate.
3. Summary of Significant Accounting Policies
Principles of Consolidation
The accompanying consolidated financial statements are prepared in accordance with United States generally accepted accounting principles (“GAAP”) and include the financial statements of RespireRx and its wholly owned subsidiary, Pier. Intercompany balances and transactions have been eliminated in consolidation.
Use of Estimates
The preparation of financial statements in conformity with GAAP requires management to make estimates and assumptions. These estimates and assumptions affect the reported amounts of assets and liabilities, disclosure of contingent assets and liabilities at the date of the financial statements and the reported amounts of revenues and expenses during the reporting period. Significant estimates include, among other things, accounting for potential liabilities, and the assumptions used in valuing stock-based compensation issued for services. Actual amounts may differ from those estimates.
Reverse Stock Split on January 5, 2021
On January 5, 2021, the Company effected a ten to one reverse-stock split of its common stock. Every ten shares of the “old” common stock was exchanged for one “new” share of common stock rounded down to the nearest whole share with any fractional shares of common stock paid to the stockholder in cash. Option and warrant issuances prior to January 5, 2021 have also been proportionately adjusted by dividing the number of shares into which such options and warrants may exercise by ten and multiplying the exercise price by ten. The effect of the reverse-stock split has been reflected retroactively in the Company’s consolidated financial statements as of December 31, 2020 and 2019 and for the fiscal years ended December 31, 2020 and 2019.
Concentrations of Credit Risk
Financial instruments that potentially subject the Company to concentrations of credit risk consist primarily of cash and cash equivalents. The Company limits its exposure to credit risk by investing its cash with high quality financial institutions. The Company’s cash balances may periodically exceed federally insured limits. The Company has not experienced a loss in such accounts to date.
Value of Financial Instruments
The authoritative guidance with respect to fair value of financial instruments established a fair value hierarchy that prioritizes the inputs to valuation techniques used to measure fair value into three levels and requires that assets and liabilities carried at fair value be classified and disclosed in one of three categories, as presented below. Disclosure as to transfers into and out of Levels 1 and 2, and activity in Level 3 fair value measurements, is also required.
Level 1. Observable inputs such as quoted prices in active markets for an identical asset or liability that the Company has the ability to access as of the measurement date. Financial assets and liabilities utilizing Level 1 inputs include active-exchange traded securities and exchange-based derivatives.
Level 2. Inputs, other than quoted prices included within Level 1, which are directly observable for the asset or liability or indirectly observable through corroboration with observable market data. Financial assets and liabilities utilizing Level 2 inputs include fixed income securities, non-exchange-based derivatives, mutual funds, and fair-value hedges.
| F-12 |
Level 3. Unobservable inputs in which there is little or no market data for the asset or liability which requires the reporting entity to develop its own assumptions. Financial assets and liabilities utilizing Level 3 inputs include infrequently traded, non-exchange-based derivatives and commingled investment funds, and are measured using present value pricing models.
The Company determines the level in the fair value hierarchy within which each fair value measurement falls in its entirety, based on the lowest level input that is significant to the fair value measurement in its entirety. In determining the appropriate levels, the Company performs an analysis of the assets and liabilities at each reporting period end.
The carrying amounts of financial instruments (consisting of cash and accounts payable and accrued expenses) are considered by the Company to be representative of the respective fair values of these instruments due to the short-term nature of those instruments. With respect to the note payable to SY Corporation and the convertible notes payable, management does not believe that the credit markets have materially changed for these types of borrowings since the original borrowing date. The Company considers the carrying amounts of the notes payable to officers, inclusive of accrued interest, to be representative of the respective fair values of such instruments due to the short-term nature of those instruments and their terms.
Deferred Financing Costs
Costs incurred in connection with ongoing debt and equity financings, including legal fees, are deferred until the related financing is either completed or abandoned.
Costs related to abandoned debt or equity financings are charged to operations in the period of abandonment. Costs related to completed equity financings are netted against the proceeds.
Capitalized Financing Costs
The Company presents debt issuance costs related to debt liability in its consolidated balance sheet as a direct deduction from the carrying amount of that debt liability, consistent with the presentation for debt discounts.
Convertible Notes Payable
Convertible notes are evaluated to determine if they should be recorded at amortized cost. To the extent that there are associated warrants, commitment shares or a beneficial conversion feature, the convertible notes and warrants are evaluated to determine if there are embedded derivatives to be identified, bifurcated and valued at fair value in connection with and at the time of such financing.
Notes Exchanges
In cases where debt or other liabilities are exchanged for equity, the Company compares the carrying value of debt, inclusive of accrued interest, if applicable, being exchanged, to the value of the equity issued and records any loss or gain as a result of such exchange. See Note 4. Notes Payable.
Extinguishment of Debt and Settlement of Liabilities
The Company accounts for the extinguishment of debt and settlement of liabilities by comparing the carrying value of the debt or liability to the value of consideration paid or assets given up and recognizing a loss or gain in the consolidated statement of operations in the amount of the difference in the period in which such transaction occurs.
| F-13 |
Prepaid Insurance
Prepaid insurance represents the premium paid in March 2020 for directors’ and officers’ insurance as well as the amount paid in April 2020 for office-related insurances and clinical trial coverage. Directors’ and Officers’ insurance tail coverage, purchased in March 2013 expired in March 2020 and all prepaid amounts have been fully amortized. The amounts of prepaid insurance amortizable in the ensuing twelve-month period are recorded as prepaid insurance in the Company’s consolidated balance sheet at each reporting date and amortized to the Company’s consolidated statement of operations for each reporting period.
Stock-Based Awards
The Company periodically issues common stock and stock options to officers, directors, Scientific Advisory Board members, consultants and other vendors for services rendered. Such issuances vest and expire according to terms established at the issuance date of each grant.
The Company accounts for stock-based payments to officers and directors, outside consultants and vendors measuring the cost of services received in exchange for equity awards based on the grant date fair value of the awards, with the cost recognized as compensation expense on the straight-line basis in the Company’s consolidated financial statements over the vesting period of the awards.
Stock grants, which are sometimes subject to time-based vesting, are measured at the grant date fair value of the common stock and charged to operations ratably over the vesting period.
Stock options granted to members of the Company’s outside consultants and other vendors are valued on the grant date. As the stock options vest, the Company recognizes this expense over the period in which the services are provided.
The value of stock options granted as stock-based compensation is determined utilizing the Black-Scholes option-pricing model, and is affected by several variables, the most significant of which are the life of the equity award, the exercise price of the stock option as compared to the fair value of the common stock on the grant date, and the estimated volatility of the common stock over the estimated life of the equity award. Estimated volatility is based on the historical volatility of the Company’s common stock. The risk-free interest rate is based on the U.S. Treasury yield curve in effect at the time of grant. The fair value of common stock is determined by reference to the quoted market price of the Company’s common stock.
Stock options and warrants issued to non-employees as compensation for services to be provided to the Company or in settlement of debt are accounted for based upon the fair value of the services provided or the estimated fair value of the stock option or warrant, whichever can be more clearly determined. Management uses the Black-Scholes option-pricing model to determine the fair value of the stock options and warrants issued by the Company. The Company recognizes this expense over the period in which the services are provided.
As of December 31, 2020, there were stock option grants exercisable into 6,750,000 shares of common stock granted to one officer who is also a director, one director who is not an officer, consultants and other vendors. Certain stock options granted were subject to vesting schedules. Stock options exercisable into 5,825,000 shares of common stock vested during the fiscal year ended December 31, 2020. The Black Scholes value of vested stock options granted during the fiscal year ended December 31, 2020 was $384,250. During the fiscal year ended December 31, 2019, there were no stock options granted to officers, directors, Scientific Advisory Board members, consultants or other vendors.
| F-14 |
For stock options requiring an assessment of fair value during the fiscal years ended December 31, 2020 and 2019 the fair value of each stock option award was estimated using the Black-Scholes option-pricing model using the following assumptions:
| 2020 | 2019 | |||||||
| Risk-free interest rate | 0.21-0.28 | % | - | % | ||||
| Expected dividend yield | 0 | % | - | % | ||||
| Expected volatility | 412.81-426.92 | % | - | % | ||||
| Expected life at date of issuance | 5 | - | ||||||
The expected life is estimated to be equal to the term of the common stock options issued in 2020.
The Company recognizes the fair value of stock-based awards in general and administrative costs and in research and development costs, as appropriate, in the Company’s consolidated statements of operations. The Company issues new shares of common stock to satisfy stock option and warrant exercises. There were no stock options exercised during the fiscal years ended December 31, 2020 and 2019.
There were no warrants issued as compensation or for services during the fiscal years ended December 31, 2020 and 2019 requiring such assessment. Warrants, if issued for services, are typically issued to placement agents or brokers for fund raising services and are not issued from any of the Company’s stock and option plans, from which options issued to non-employees for services are typically issued.
Income Taxes
The Company accounts for income taxes under an asset and liability approach for financial accounting and reporting for income taxes. Accordingly, the Company recognizes deferred tax assets and liabilities for the expected impact of differences between the financial statements and the tax basis of assets and liabilities.
The Company records a valuation allowance to reduce its deferred tax assets to the amount that is more likely than not to be realized. In the event the Company was to determine that it would be able to realize its deferred tax assets in the future in excess of its recorded amount, an adjustment to the deferred tax assets would be credited to operations in the period such determination was made. Likewise, should the Company determine that it would not be able to realize all or part of its deferred tax assets in the future, an adjustment to the deferred tax assets would be charged to operations in the period such determination was made.
Pursuant to Internal Revenue Code Sections 382 and 383, use of the Company’s net operating loss and credit carryforwards may be limited if a cumulative change in ownership of more than 50% occurs within any three-year period since the last ownership change. The Company may have had a change in control under these Sections. However, the Company does not anticipate performing a complete analysis of the limitation on the annual use of the net operating loss and tax credit carryforwards until the time that it anticipates it will be able to utilize these tax attributes.
As of December 31, 2020, the Company did not have any unrecognized tax benefits related to various federal and state income tax matters and does not anticipate any material amount of unrecognized tax benefits within the next 12 months.
The Company is subject to U.S. federal income taxes and income taxes of various state tax jurisdictions. As the Company’s net operating losses have yet to be utilized, all previous tax years remain open to examination by Federal authorities and other jurisdictions in which the Company currently operates or has operated in the past.
The Company accounts for uncertainties in income tax law under a comprehensive model for the financial statement recognition, measurement, presentation and disclosure of uncertain tax positions taken or expected to be taken in income tax returns as prescribed by GAAP. The tax effects of a position are recognized only if it is “more-likely-than-not” to be sustained by the taxing authority as of the reporting date. If the tax position is not considered “more-likely-than-not” to be sustained, then no benefits of the position are recognized. As of December 31, 2020, the Company had not recorded any liability for uncertain tax positions. In subsequent periods, any interest and penalties related to uncertain tax positions will be recognized as a component of income tax expense.
| F-15 |
Foreign Currency Transactions
The note payable to SY Corporation, which is denominated in a foreign currency (the South Korean Won), is translated into the Company’s functional currency (the United States Dollar) at the exchange rate on the balance sheet date. The foreign currency exchange gain or loss resulting from translation is recognized in the related consolidated statements of operations.
Research and Development
Research and development costs include compensation paid to management directing the Company’s research and development activities, including but not limited to compensation paid to our Chief Scientific Officer and fees paid to consultants and outside service providers and organizations (including research institutes at universities), and other expenses relating to the acquisition, design, development and clinical testing of the Company’s treatments and product candidates.
License Agreements
Obligations incurred with respect to mandatory payments provided for in license agreements are recognized ratably over the appropriate period, as specified in the underlying license agreement, and are recorded as liabilities in the Company’s consolidated balance sheet, with a corresponding charge to research and development costs in the Company’s consolidated statement of operations. Obligations incurred with respect to milestone payments provided for in license agreements are recognized when it is probable that such milestone will be reached and are recorded as liabilities in the Company’s consolidated balance sheet, with a corresponding charge to research and development costs in the Company’s consolidated statement of operations.
Patent Costs
Due to the significant uncertainty associated with the successful development of one or more commercially viable products based on the Company’s research efforts and any related patent applications, all patent costs, including patent-related legal and filing fees, are expensed as incurred and are charged to general and administrative expenses.
Earnings per Share
The Company’s computation of earnings per share (“EPS”) includes basic and diluted EPS. Basic EPS is measured as the income (loss) attributable to common stockholders divided by the weighted average common shares outstanding for the period. Diluted EPS is similar to basic EPS but presents the dilutive effect on a per share basis of potential common shares (e.g., warrants and options) as if they had been converted at the beginning of the periods presented, or issuance date, if later. Potential common shares that have an anti-dilutive effect (i.e., those that increase income per share or decrease loss per share) are excluded from the calculation of diluted EPS.
| F-16 |
Net income (loss) attributable to common stockholders consists of net income or loss, as adjusted for actual and deemed preferred stock dividends declared, amortized or accumulated. The Company recorded a deemed dividend for the issuance of warrants during year ended December 31, 2020 of $1,440,214. The deemed dividend is added to the net loss in determining the net loss available to common stockholders.
Loss per common share is computed by dividing net loss by the weighted average number of shares of common stock outstanding during the respective periods. Basic and diluted loss per common share is the same for all periods presented because all warrants and stock options outstanding are anti-dilutive.
At December 31, 2020 and 2019, the Company excluded the outstanding securities summarized below, which entitle the holders thereof to acquire shares of common stock, from its calculation of earnings per share, as their effect would have been anti-dilutive.
| December 31, | ||||||||
| 2020 | 2019 | |||||||
| Series B convertible preferred stock | 1 | 11 | ||||||
| Convertible notes payable | 13,333,036 | 7,017,896 | ||||||
| Common stock warrants | 28,809,352 | 2,191,043 | ||||||
| Common stock options | 7,165,215 | 4,344,994 | ||||||
| Total | 49,307,604 | 13,553,944 | ||||||
Reclassifications
Certain comparative figures in 2019 have been reclassified to conform to the current year’s presentation. These reclassifications were immaterial, both individually and in the aggregate.
Recent Accounting Pronouncements
In August 2020, the FASB issued Accounting Standards Update No. 2020-06, Debt – Debt with Conversion and Other Options (Subtopic 470-20) and Derivatives and Hedging—Contracts in Entity’s Own Equity (Subtopic 815-40). The subtitle is Accounting for Convertible Instruments and Contracts in an Entity’s Own Equity. This Accounting Standard Update (“ASU”) addresses complex financial instruments that have characteristics of both debt and equity. The application of this ASU would reduce the number of accounting models for convertible debt instruments and convertible preferred stock. Limiting the accounting models would result in fewer embedded conversion features being separately recognized from the host contract as compared with current GAAP. Convertible instruments that continue to be subject to separation models are (1) those with embedded conversion features that are not clearly and closely related to the host contract, that meet the definition of a derivative, and that do not qualify for a scope exception from derivative accounting and (2) convertible debt instruments issued with substantial premiums for which the premiums are recorded as paid-in capital. The Company has historically issued complex financial instruments and has considered whether embedded conversion features have existed within those contracts or whether derivatives would appropriately be bifurcated. To date, no such bifurcation has been necessary. However, it is possible that this ASU may have a substantial impact on the Company’s financial statements. Management is evaluating the potential impact. This ASU becomes effective for fiscal years beginning after December 15, 2023.
In January 2020, the FASB issued ASU 2020-01, Clarifying the Interactions between Topic 321, Topic 323, Equity Method and Joint Ventures, and Topic 815, Derivatives and Hedging which represents an amendment clarifying the interaction between accounting standards related to equity securities, equity method investments and certain derivatives. The guidance is effective for fiscal years beginning after December 15, 2020. Management is currently evaluating the impact the guidance will have on our consolidated financial statements.
In August 2018, the FASB issued ASU 2018-13, Disclosure Framework-Changes to the Disclosure Requirements for Fair Value Measurement, an accounting standard update which modifies the disclosure requirements on fair value measurements. This guidance was effective for fiscal years beginning after December 15, 2019. The amendments related to the range and weighted average of significant unobservable inputs used to develop Level 3 fair value measurements, and the narrative description of measurement uncertainty was to be applied prospectively. All other amendments were to be applied retrospectively. An entity was permitted to early adopt any removed or modified disclosures upon issuance of this guidance and delay adoption of the additional disclosures until their effective date. The adoption of this guidance did not have a material impact on our consolidated financial statements in the year ended December 31, 2020.
| F-17 |
4. Notes Payable
Convertible Notes Payable
Convertible Note with EMA Financial, LLC
On July 30, 2020, RespireRx and EMA Financial, LLC (“EMA”) entered into a Securities Purchase Agreement (the “EMA SPA”) by which EMA provided a sum of $68,250 (the “EMA Consideration”) to RespireRx, in return for a fixed rate convertible note (the “EMA Note”) with a face amount of $75,000, and a common stock purchase warrant (the “EMA Warrant”) for 375,000 shares of Common Stock (post-reverse stock split basis). The net proceeds received by RespireRx on August 4, 2020 were $63,750 after payment of $3,500 in EMA’s legal fees and the withholding by EMA of $1,000 in diligence fees.
The EMA Note obligates RespireRx to pay by October 30, 2021 (the “EMA Maturity Date”) a principal amount of $75,000 together with interest at a rate equal to 10% per annum, which principal exceeds the EMA Consideration by the amount of an original issue discount of $6,750. Any amount of principal or interest that is not paid by the EMA Maturity Date would bear interest at the rate of 24% from the EMA Maturity Date to the date such amount is paid.
EMA has the right, in its discretion, at any time, to convert any outstanding and unpaid amount of the EMA Note into shares of Common Stock, provided that such conversion would not result in EMA beneficially owning more than 4.99% of RespireRx’s then outstanding Common Stock. In the absence of an event of default, EMA may convert at a per share conversion price equal to $0.02, subject to a retroactive downward adjustment if the lowest traded price on each of the three consecutive trading days following such conversion is lower than $0.02. Upon an event of default, the conversion price is adjusted downward based on a discount to market with respect to subsequent financings or a percentage of the lowest traded price during the twenty-one-day period prior to the conversion, if lower than $0.02. Upon such conversion, all rights with respect to the portion of the EMA Note being so converted terminate, except for the right to receive Common Stock or other securities, cash or other assets as provided in the EMA Note due upon such conversion.
RespireRx may, with prior written notice to EMA, prepay the outstanding principal amount under the EMA Note during the initial 180 day period by making a payment to EMA of an amount in cash equal to a certain percentage of the outstanding principal, interest, default interest and other amounts owed. Such percentage varies from 110% to 115% depending on the period in which the prepayment occurs, as set forth in the EMA Note.
If, prior to the repayment or conversion of the EMA Note, RespireRx consummates a registered, qualified or unregistered primary offering of its securities for capital raising purposes with aggregate net proceeds in excess of $2,500,000, EMA will have the right, in its discretion, to demand repayment in full of any outstanding principal, interest (including default interest) under the EMA Note as of the closing date of such offering.
The EMA SPA includes, among other things: (1) an automatic adjustment to the terms of the EMA SPA and related documents to the terms of a future financing if those terms are more beneficial to an investor than the terms of the EMA SPA and related documents are to EMA, subject to limited exceptions; and (2) certain registration rights. In addition, the EMA Note prohibits RespireRx from selling or otherwise disposing of a significant portion of its assets outside the ordinary course of business or in connection with a merger or consolidation or sale of all or substantially all of RespireRx’s assets where the surviving or successor entity does not assume RespireRx’s obligations under the EMA SPA. Further, any subsidiary to which RespireRx transfers a material amount of assets must guarantee certain obligations of RespireRx under the EMA Note.
The EMA Warrant is a common stock purchase warrant to purchase 375,000 shares of Common, for value received in connection with the issuance of the EMA Note, from the date of issuance of the EMA Warrant until September 30, 2023, at an exercise price of $0.07 (subject to adjustment as provided therein) per share of Common Stock.
| F-18 |
The Company determined that there were no embedded derivatives to be identified, bifurcated and valued in connection with this financing.
The outstanding amounts of the EMA Note consists of the following at December 31, 2020 and December 31, 2019:
December 31, 2020 | December 31, 2019 | |||||||
| Principal amount of notes payable | $ | 75,000 | $ | - | ||||
| Unamortized portion of note discounts | (18,417 | ) | - | |||||
| Accrued interest payable | 3,164 | - | ||||||
| $ | 59,747 | $ | - | |||||
Convertible Note and Equity Purchase Agreement with White Lion Capital, LLC
On July 28, 2020, RespireRx issued a convertible note, as amended (“Commitment Note”) to White Lion Capital, LLC (“White Lion”) pursuant to, and to induce White Lion to enter into an equity purchase agreement dated July 28, 2020 (“White Lion EPA”). See Note 9. Commitments and Contingencies - Entry into Equity Purchase Agreement for a description of the White Lion EPA and the other agreements entered into pursuant to the White Lion EPA. The Commitment Note had an initial face amount of $25,000 which was subsequently amended effective July 28, 2020 to $40,000 in consideration for an amendment to the White Lion EPA extending the date by which RespireRx was to file a registration statement on Form S-1 listing White Lion as the selling stockholder on Form S-1. The Commitment Note was accounted for as equity issuance costs in Additional paid-in capital.
The Commitment Note obligates RespireRx to pay by July 28, 2021 a principal amount of $40,000, together with a guaranteed interest payment of $3,200 representing an 8% per annum interest rate applied regardless of any payments or prepayments other than payments made by conversion of the Commitment Note. Upon an event of default, any amount of outstanding principal or interest would bear interest at the lower of 18% or the highest rate permitted by law.
White Lion has the right, at any time after the first 180 days after execution of the White Lion EPA, to convert any outstanding and unpaid amount (including accrued interest and other fees) into shares of Common Stock, provided that such conversion would not result in White Lion beneficially owning more than 9.99% of the Company’s then outstanding Common Stock. Unless an event of default has occurred, White Lion may convert at a per share conversion price equal to $0.02. Upon such conversion, all rights with respect to the portion of the Commitment Note being so converted terminate, except for the right to receive Common Stock. White Lion also has the right, at any time the Commitment Note is outstanding, to apply any outstanding principal or interest as consideration for any equity, equity-linked and/or debt securities offered by the Company in any public offering or private placement, subject to the terms of the Commitment Note.
RespireRx may, with prior written notice to White Lion, prepay the entire outstanding principal amount under the Commitment Note at any time by making a payment to White Lion of an amount in cash equal to 110% of the outstanding principal, guaranteed interest amount, and any default interest or other amounts owed.
RespireRx determined that there were no embedded derivatives to be identified, bifurcated and valued in connection with this financing.
The outstanding amounts of the White Lion Note consist of the following at December 31, 2020 and December 31, 2019:
December 31, 2020 | December 31, 2019 | |||||||
| Principal amount of notes payable | $ | 40,000 | $ | - | ||||
| Accrued interest payable | 1,368 | - | ||||||
| $ | 41,368 | $ | - | |||||
| F-19 |
Convertible Note with FirstFire Global Opportunities Fund LLC
On July 2, 2020, RespireRx and FirstFire Global Opportunities Fund LLC (“FirstFire”) entered into a Securities Purchase Agreement (the “FirstFire SPA”) pursuant to which FirstFire provided a sum of $125,000 (the “FirstFire Consideration”) to RespireRx, in return for a convertible promissory note (the “FirstFire Note”) with a face amount of $137,500 (which difference in value as compared to the FirstFire Consideration is due to an original issue discount of $12,500), a common stock purchase warrant for 687,500 shares of the Company’s common stock (post-reverse stock split basis) (the “FirstFire Warrant”), and the Confession of Judgment (as defined below), among other agreements and obligations. The net proceeds of the First Fire Consideration, which were received by RespireRx on July 6, 2020, equal $121,000 after payment of $4,000 in FirstFire’s legal fees.
Under the terms of the FirstFire SPA and the FirstFire Note, FirstFire paid the FirstFire Consideration at closing. The FirstFire Note obligates RespireRx to pay interest at a rate of 10% per annum on any unpaid principal beginning on July 2, 2020, and to make five monthly amortization payments in the amount of $30,250 each, with the first such payment due on December 2, 2020, and the final such payment, along with any unpaid principal and any accrued and unpaid interest and other fees, due April 2, 2021 (the “FirstFire Note Maturity Date”). Any amount of principal or interest that is not paid when due bears interest at the rate of the lesser of 24% and the maximum amount permitted by law, from the due date to the date such amount is paid.
FirstFire has the right, at any time, to convert any outstanding and unpaid amount of the FirstFire Note into shares of RespireRx’s Common Stock or securities convertible into RespireRx’s common stock, provided that such conversion would not result in FirstFire beneficially owning more than 4.99% of RespireRx’s then outstanding shares of Common Stock. Subject to certain limitations and adjustments as described in the FirstFire Note, FirstFire may convert at a per share conversion price equal to $0.02 (the “FirstFire Fixed Conversion Price”), provided that upon any event of default, the conversion price will equal the lower of (i) the FirstFire Fixed Conversion Price, (ii) discount to market based upon subsequent financings with other investors, or (iii) 60% multiplied by the lowest traded price of Common Stock during the twenty-one consecutive trading day period immediately preceding the date of such conversion. Upon such conversion, all rights with respect to the portion of the FirstFire Note being so converted terminate, except for the right to receive Common Stock or other securities, cash or other assets as provided in the FirstFire Note due upon such conversion.
RespireRx may, with prior written notice to FirstFire, prepay the outstanding principal amount under the FirstFire Note during the initial 180 day period after the execution of the FirstFire SPA by making a payment to FirstFire of an amount in cash equal to a certain percentage of the outstanding principal, interest, default interest and other amounts owed. Such percentage varies from 105% to 115% depending on the period in which the prepayment occurs.
The FirstFire SPA provides FirstFire with certain participation rights in any subsequent offering of debt or equity. Under the FirstFire SPA, RespireRx may not enter into an offering of its securities with terms that would benefit an investor more than FirstFire is benefited under the FirstFire SPA and the agreements ancillary thereto, unless RespireRx offers FirstFire those same terms. The FirstFire SPA also grants FirstFire certain registration rights.
The FirstFire Warrant is a warrant to purchase 687,500 shares of Common Stock, for value received in connection with the issuance of the FirstFire Note, from the date of issuance of the FirstFire Warrant until September 30, 2023, at an exercise price of $0.07 (subject to adjustment as provided therein) per share of common stock.
Additionally, RespireRx provided a confession of judgment (the “Confession of Judgment”) in favor of FirstFire for the amount of the FirstFire Note plus fees and costs, to be filed pursuant to the terms and conditions of the FirstFire SPA and the FirstFire Note.
The Company determined that there were no embedded derivatives to be identified, bifurcated and valued in connection with this financing.
| F-20 |
The outstanding amounts of the FirstFire Note consist of the following at December 31, 2020 and December 31, 2019:
December 31, 2020 | December 31, 2019 | |||||||
| Principal amount of notes payable | $ | 137,500 | $ | - | ||||
| Unamortized portion of note discounts | (14,916 | ) | - | |||||
| Accrued interest payable | 6,856 | - | ||||||
| $ | 129,440 | $ | - | |||||
Convertible Notes with PowerUp Lending Group Ltd.
RespireRx and PowerUp Lending Group Ltd. (“PowerUp”) entered into two securities purchase agreements, dated as of April 15, 2020 and June 7, 2020 (each, a “PowerUp Agreement”), by which Power Up loaned $53,000 and $43,000, respectively, to RespireRx in return for two convertible promissory notes (the “April 2020 Note” and the “June 2020 Note” respectively), a limited guaranty associated with the April 2020 Note, and the delivery into escrow of a confession of judgment in favor of Power Up for the amount of the April 2020 Note plus fees and costs to be filed by Power Up upon the occurrence of an Event of Default (as defined in the April 2020 Note) and other transaction-related documents associated with both the April 2020 Note and the June 2020 Note. The proceeds of the loans, which equal $90,000 after payment of $5,000 in legal fees and $1,000 in due diligence fees, were used for general corporate purposes.
The April 2020 Note was repaid by conversion in October 2020. The June 2020 Note which would have payable on June 7, 2021, (the “June 2020 Note Maturity Date”), and bears interest at a rate equal to 12% per annum, with any amount of principal or interest which is not paid when due bearing interest at the rate of 22% per annum was paid in full in December 2020.
There were no outstanding amounts due with respect to the April 2020 Note and June 2020 Note as of December 31, 2020.
On October 22, 23 and 26, 2020, Power Up converted the outstanding principal amount and all accrued and unpaid interest related to the April 2020 Note into 2,080,740 shares of Common Stock and as of October 26, 2020 the April 2020 Note is deemed repaid and terminated. On December 14, 2020 and December 15, 2020, converted the outstanding principal amount and all accrued and unpaid interest related to the June 2020 Note into 3,506,153 shares of Common Stock (post-reverse stock split basis) and the June 2020 Note is deemed repaid and terminated.
2019 Convertible Notes
On April, 24, 2019, May 17, 2019, August 19, 2019, October 22, 2019 and November 4, 2019, the Company issued a series of convertible notes (“2019 Convertible Notes”), all similar in nature, all subject to debt issuance costs (“DIC”) and original issue discount (“OID”) and beneficial conversion (“BCF”) features and some subject to the issuance of warrants (“NW”) and/or commitment shares (“CS”) and placement agent fees. Two of the notes had maturity dates nine months after issuance and three were for one year. One note was a master note agreement in the amount of $150,000, but with an initial drawdown of $50,000. The Company evaluated all of the terms of the 2019 Convertible Notes and determined that, in accordance with ASC 815, there were no derivatives to be bifurcated or separately valued. Each of the April, 24, 2019, May 17, 2019, August 19, 2019, October 22, 2019 and November 4, 2019 Convertible Notes was satisfied in full by the lenders electing to convert the outstanding balances to Common Stock, except for $2,747 of accrued interest that remains outstanding under the May 17, 2019 Convertible Note.
| Inception date | Maturity date | Original principal amount | Interest rate | Original aggregate DIC, OID, BCF, NW and CS | Cumulative amortization of DIC, OID, BCF, NW and CS | Principal remaining at December 31, 2020 | Accrued Interest at December 31, 2020 | Balance sheet carrying amount at December 31, 2020 inclusive of accrued interest | ||||||||||||||||||||||
| May 17, 2019 | May 17, 2020, extended to November 17, 2020 | $ | 50,000 | 10 | % | $ | 50,000 | $ | 50,000 | $ | - | $ | 2,747 | $ | 2,747 | |||||||||||||||
| Total | $ | 50,000 | $ | 50,000 | $ | 50,000 | $ | - | $ | 2,747 | $ | 2,747 | ||||||||||||||||||
| F-21 |
On December 6, 2018, December 7, 2018 and December 31, 2018 the Company issued convertible notes (each a “2018 Q4 Note”) and on January 2, 2019, February 27, 2019, March 6, 2019 and March 14, 2019, the Company issued additional convertible notes (each a “2019 Q1 Note”, respectively and collectively with the “2018 Q4, the “2018 Q4 and 2019 Q1 Notes”) bearing interest at 10% per year. All of the 2018 Q4 and 2019 Q1 Notes matured on either February 28, 2019 or April 30, 2019. The original aggregate principal amount was $190,000. None of the 2018 Q4 and 2019 Q1 Notes were repaid at maturity. The 2018 Q4 and 2019 Q1 Note investors also received an aggregate of 19,000 common stock purchase warrants. The warrants were valued using the Black Scholes option pricing model calculated on the date of each grant and had an aggregate value of $146,805. Total value received by the investors was $336,805, the sum of the face value of the convertible note and the value of the warrant. Therefore, the Company recorded a debt discount associated with the warrant issuance of $82,159 and an initial value of the convertible notes of $107,841 using the relative fair value method. All debt discounts were fully amortized by the original maturity dates. On March 21, 2020, all except one of the 2018 Q4 and 2019 Q1 Note holders exchanged the outstanding principal amount and accrued interest for shares of common stock. The exchange price was $0.15 per share of common stock. The closing price on March 20, 2020, the last trading day before the closing of the exchange agreements which took place on a Saturday, was $0.34 per share of common stock. An aggregate of $155,000 of principal and $17,911 of accrued interest was exchanged for 1,152,740 shares of common stock. The Company recorded a loss on the extinguishment of the exchanged 2018 Q4 Notes and 2019 Q1 Notes of $219,021. As of December 31, 2020, there remains one outstanding 2018 Q4 Note and one outstanding 2019 Q1 Note, both held by the same single investor, with an aggregate principal amount of $35,000 and aggregate accrued interest of $7,201 as of December 31, 2020. The 2019 Convertible Notes discussed above, which the Company does not consider to have arisen from one or more offerings, may be interpreted in such a way that the remaining 2018 Q4 Note and 2019 Q1 Note holders had the right to convert or exchange into such notes. However, no holder of the Q4 2018 and 2019 Notes has requested such a conversion or exchange. The Company does not believe that an offering occurred as of December 31, 2020 or as of the date of the issuance of these financial statements. Therefore, the number of shares of common stock (or preferred stock) into which the remaining 2018 Q4 Note and the remaining 2019 Q1 Note may convert is not determinable and the Company has not accounted for any additional consideration. The warrants to purchase 19,000 shares of common stock issued in connection with the sale of the 2018 Q4 and 2019 Q1 Notes are exercisable at a fixed price of $15.00 per share of common stock, provide no right to receive a cash payment, and included no reset rights or other protections based on subsequent equity transactions, equity-linked transactions or other events. The warrants issued to the Q4 2018 and Q1 2019 Note holders expire on December 30, 2023. The Company determined that there were no embedded derivatives to be identified, bifurcated and valued in connection with this financing.
The 2018 Q4 Notes and 2019 Q1 Notes consist of the following at December 31, 2020 and December 31, 2019:
December 31, 2020 | December 31, 2019 | |||||||
| Principal amount of notes payable | $ | 35,000 | $ | 190,000 | ||||
| Accrued interest payable | 7,201 | 17,976 | ||||||
| $ | 42,201 | $ | 207,976 | |||||
| F-22 |
Other convertible notes were also sold to investors in 2014 and 2015 (the “Original Convertible Notes), which aggregated a total of $579,500, and had a fixed interest rate of 10% per annum. The Original Convertible Notes have no reset rights or other protections based on subsequent equity transactions, equity-linked transactions or other events. The warrants to purchase shares of common stock issued in connection with the sale of the Original Convertible Notes have either been exchanged for common stock or expired.
On March 21, 2020, the holder of one of the Original Convertible Notes exchanged $50,000 of principal and $32,875 of accrued interest for 552,501 shares of the Company’s common stock. The exchange price was $0.15 per share of common stock. The closing price on March 20, 2020, the last trading day before the closing of the exchange agreements, was $0.34 per share of common stock. The Company recorded a loss on the extinguishment of the exchanged Original Convertible Note of $104,975.
The remaining outstanding Original Convertible Notes (including that for which a default notice has been received) consist of the following at December 31, 2020 and December 31, 2019:
December 31, 2020 | December 31, 2019 | |||||||
| Principal amount of notes payable | $ | 75,000 | $ | 125,000 | ||||
| Accrued interest payable | 64,357 | 82,060 | ||||||
| $ | 139,357 | $ | 207,060 | |||||
As of December 31, 2020, principal and accrued interest on the Original Convertible Note that is subject to a default notice accrues annual interest at 12% instead of 10%, totaled $48,700, of which $23,700 was accrued interest. As of December 31, 2019, principal and accrued interest on Original Convertible Notes subject to default notices totaled $43,666 of which $18,666 was accrued interest.
As of December 31, 2020 all of the outstanding Original Convertible Notes, inclusive of accrued interest, were convertible into an aggregate of 1,225 shares of the Company’s common stock (post-reverse stock split basis). Such Original Convertible Notes will continue to accrue interest until exchanged, paid or otherwise discharged. There can be no assurance that any of the additional holders of the remaining Original Convertible Notes will exchange their Original Convertible Notes.
Note Payable to SY Corporation Co., Ltd.
On June 25, 2012, the Company borrowed 465,000,000 Won (the currency of South Korea, equivalent to approximately $400,000 United States Dollars) from and executed a secured note payable to SY Corporation Co., Ltd., formerly known as Samyang Optics Co. Ltd. (“SY Corporation”), an approximately 20% common stockholder of the Company at that time. SY Corporation was a significant stockholder and a related party at the time of the transaction, but has not been a significant stockholder or related party of the Company subsequent to December 31, 2014. The note accrues simple interest at the rate of 12% per annum and had a maturity date of June 25, 2013. The Company has not made any payments on the promissory note. At June 30, 2013 and subsequently, the promissory note was outstanding and in default, although SY Corporation has not issued a notice of default or a demand for repayment. The Company believes that SY Corporation is in default of its obligations under its January 2012 license agreement, as amended, with the Company, but the Company has not yet issued a notice of default. The Company intends to continue efforts towards a comprehensive resolution of the aforementioned matters involving SY Corporation.
| F-23 |
The promissory note is secured by collateral that represents a lien on certain patents owned by the Company, including composition of matter patents for certain of the Company’s high impact ampakine compounds and the low impact ampakine compounds CX2007 and CX2076, and other related compounds. The security interest does not extend to the Company’s patents for its ampakine compounds CX1739 and CX1942, or to the patent for the use of ampakine compounds for the treatment of respiratory depression.
Note payable to SY Corporation consists of the following at December 31, 2020 and 2019:
| December 31, 2020 | December 31, 2019 | |||||||
| Principal amount of note payable | $ | 399,774 | $ | 399,774 | ||||
| Accrued interest payable | 411,384 | 363,280 | ||||||
| Foreign currency transaction adjustment | 53,393 | 3,182 | ||||||
| $ | 864,551 | $ | 766,236 | |||||
Interest expense with respect to this promissory note was $48,104 and $47,971 for years ended December 31, 2020 and 2019, respectively.
Advances from and Notes Payable to Officers
On January 29, 2016, Dr. Arnold S. Lippa, the Company’s Interim President, Interim Chief Executive Officer, Chief Scientific Officer and Chairman of the Board of Directors, advanced $52,600 to the Company for working capital purposes under a demand promissory note with interest at 10% per annum. On September 23, 2016, Dr. Lippa advanced $25,000 to the Company for working capital purposes under a second demand promissory note with interest at 10% per annum. The notes are secured by the assets of the Company. Additionally, on April 9, 2018, Dr. Lippa advanced another $50,000 to the Company as discussed in more detail below.
On February 2, 2016, Dr. James S. Manuso, the Company’s then Chief Executive Officer and Vice Chairman of the Board of Directors, advanced $52,600 to the Company for working capital purposes under a demand promissory note with interest at 10% per annum. On September 22, 2016, Dr. Manuso, advanced $25,000 to the Company for working capital purposes under a demand promissory note with interest at 10% per annum. The notes are secured by the assets of the Company. Additionally, on April 9, 2018, Dr. Manuso advanced another $50,000 to the Company as discussed in more detail below.
On April 9, 2018, Dr. Arnold S. Lippa, the Company’s Interim President, Interim Chief Executive Officer, Chief Scientific Officer and Chairman of the Board of Directors and Dr. James S. Manuso, the Company’s then Chief Executive Officer and Vice Chairman of the Board of Directors, advanced $50,000 each, for a total of $100,000, to the Company for working capital purposes. Each note is payable on demand after June 30, 2018. Each note was subject to a mandatory exchange provision that provided that the principal amount of the note would be mandatorily exchanged into a board approved offering of the Company’s securities, if such offering held its first closing on or before June 30, 2018 and the amount of proceeds from such first closing was at least $150,000, not including the principal amounts of the notes that would be exchanged, or $250,000 including the principal amounts of such notes. Upon such exchange, the notes would be deemed repaid and terminated. Any accrued but unpaid interest outstanding at the time of such exchange will be (i) repaid to the note holder or (ii) invested in the offering, at the note holder’s election. A first closing did not occur on or before June 30, 2018. Dr. Arnold S. Lippa agreed to exchange his note into the board approved offering that had its initial closing on September 12, 2018. Accrued interest on Dr. Lippa’s note was not exchanged. As of December 31, 2020, Dr. James S. Manuso had not exchanged his note.
| F-24 |
During the year ended December 31, 2020, Dr. Lippa advanced on an interest free basis the Company $65,000 of which $16,036 was repaid to Dr. Lippa. The outstanding balance of the advance is payable on demand.
During the year ended December 31, 2020, Mr. Margolis advanced $10,775 to which when aggregated with the outstanding balance of $5,500 as of the beginning of the fiscal year, was $16,275, all of which was repaid by the Company during the fiscal year ended December 31, 2020.
For the fiscal years ended December 31, 2020 and 2019, $11,329 and $10,272 was charged to interest expense with respect to Dr. Lippa’s notes, respectively.
For the fiscal years ended December 31, 2020 and 2019, $16,988 and $15,416 was charged to interest expense with respect to Dr. James S. Manuso’s notes, respectively.
As of September 30, 2018, Dr. James S. Manuso resigned his executive officer positions and as a member of the Board of Directors of the Company. All of the interest expense noted above for 2020 and 2019 was incurred while Dr. Manuso was no longer an officer.
Other Short-Term Notes Payable
Other short-term notes payable at December 31, 2020 and December 31, 2019 consisted of premium financing agreements with respect to various insurance policies. At December 31, 2020, a premium financing agreement was payable in the initial amount of $70,762, with interest at 11% per annum, in ten monthly installments of $8,256, and another premium financing arrangement was payable in the initial amount of $9,215 payable in equal quarterly installments. At December 31, 2020 and 2019, the aggregate amount of the short-term notes payable was $4,608 and $4,635 respectively.
5. Settlement and Payment Agreements
On February 21, 2020, Sharp Clinical Services, Inc. (“Sharp”), a vendor of the Company, filed a complaint against the Company in the Superior Court of New Jersey Law Division, Bergen County related to a December 16, 2019 demand for payment of past due invoices inclusive of late fees totaling $103,890 of which $3,631 related to late fees, seeking $100,259 plus 1.5% interest per month on outstanding unpaid invoices. Amid settlement discussions, the vendor stated on March 13, 2020 its intent to proceed to a default judgment against the Company, and the Company stated on March 14, 2020 its intent to continue settlement discussions. On May 29, 2020, a default was entered against the Company, and on September 4, 2020, a final judgment by default was entered against the Company in the amount of $104,217. The Company has recorded a liability to Sharp of $103,859 as of December 31, 2020.
On February 23, 2021 our bank received two New Jersey Superior Court Levies totaling $320,911 related to amounts owed to two vendors (Sharp and Salamandra as defined below and herein) which amounts were not in dispute, debited our accounts and restricted access to those accounts. Our accounts were debited for $1,559 on February 23, 2021 which represented all of the cash in our accounts on that date.
On March 3, 2021, we executed a settlement agreement with one of the two vendors noted below (the “Sharp Settlement Agreement”). The Sharp Settlement Agreement calls for a payment schedule totaling $100,000 in ten bi-monthly installments which began on April 1, 2021 and are due every other month thereafter and permits early settlement at $75,000 if the Company pays Sharp that lower total by August 1, 2021. The first $10,000 payment that which was due on April 1, 2021, was paid on March 23, 2021. On March 9, 2021, Sharp requested of the Bergen (NJ) County Sheriff, the return of the Writ of Execution which resulted in a release of the lien.
| F-25 |
By letter dated February 5, 2016, the Company received a demand from a law firm representing Salamandra, LLC (“Salamandra”) alleging an amount due and owing for unpaid services rendered. On January 18, 2017, following an arbitration proceeding, an arbitrator awarded Salamandra the full amount sought in arbitration of $146,082. Additionally, the arbitrator granted Salamandra attorneys’ fees and costs of $47,937. All such amounts have been accrued as of December 31, 2020 and December 31, 2019, including accrued interest at 4.5% annually from February 26, 2018, the date of the judgment, through December 31, 2020, totaling $24,129.
On August 21, 2019, RespireRx and Salamandra entered into a settlement agreement and release, which was amended on December 16, 2019 (as amended, the “Salamandra Settlement Agreement”), regarding $202,395 owed by the Company to Salamandra (as reduced by any further payments by the Company to Salamandra, the “Full Amount”) in connection with the above-mentioned arbitration award. Under the Salamandra Settlement Agreement, (i) the Company paid to Salamandra $25,000 before December 21, 2019, and upon such payment, Salamandra ceased all collection efforts against the Company until March 31, 2020, and (ii) the Company was to pay to Salamandra by March 31, 2020 an amount equal to 21% of the working capital amount raised by that date, which would have reduced the Full Amount owed on a dollar-for-dollar basis. The Company did not make the required payment on March 31, 2020 and has not made any subsequent payments other than what may have been received by Salamandra pursuant to the levies described above.
On September 23, 2019, the Company and a vendor agreed in principle to a proposed settlement agreement, which has not resulted in a formal agreement. In February 2020, the Company and the vendor discussed amendments to this agreement in principal. The discussions included, among other things, an extension of time to raise the amount discussed below. The Company and the vendor are continuing discussions in an attempt to reach a formal settlement agreement.
The due date of the $100,000 annual amount payable to the University of Illinois that was originally due on December 31, 2020 pursuant to the 2014 License Agreement was extended to April 19, 2021 and was paid in full on April 1, 2021.
By email dated July 21, 2016, the Company received a demand from an investment banking consulting firm that represented the Company in 2012 in conjunction with the Pier transaction alleging that $225,000 is due and payable for investment banking services rendered. Such amount has been included in accrued expenses at December 31, 2020 and December 31, 2019.
The Company is periodically the subject of various pending and threatened legal actions and claims. In the opinion of management of the Company, adequate provision has been made in the Company’s consolidated financial statements as of December 31, 2020 and December 31, 2019 with respect to such matters, including, specifically, the matters noted above. The Company intends to vigorously defend itself if any of the matters described above results in the filing of a lawsuit or formal claim.
| F-26 |
6. Stockholders’ Deficiency
Preferred Stock
RespireRx has authorized a total of 5,000,000 shares of preferred stock, par value $0.001 per share. As of December 31, 2020 and December 31, 2019, 1,250,000 shares were designated as 9% Cumulative Convertible Preferred Stock; 37,500 shares were designated as Series B Convertible Preferred Stock (non-voting, “Series B Preferred Stock”); 205,000 shares were designated as Series A Junior Participating Preferred Stock; 1,700 shares were designated as Series G 1.5% Convertible Preferred Stock. On July 13, 2020, RespireRx designated 1,200 shares of Series H, Voting, Non-participating, Convertible Preferred Stock (“Series H Preferred Stock”) and on September 30, 2020 RespireRx amended the Certificate of Designation of the Series H Preferred Stock to increase the number of shares of Series H Preferred Stock to 3,000 shares. On July 13, 2020 and September 30, 2020, RespireRx issued an aggregate of 1,624.1552578 shares of Series H Preferred Stock inclusive of 2% accrued dividends, all of which converted on September 30, 2020 into 25,377,426 shares of Common Stock and warrants to purchase 25,377,426 shares of Common Stock, and therefore as of that time on September 30, 2020, there were no shares of Series H Preferred Stock outstanding. Under the Certificate of Designation of the Series H Preferred Stock, shares of Series H Preferred Stock converted or redeemed by conversion are to be canceled and are not to be reissued. Accordingly, on December 31, 2020 and 2019, 3,504,424.1552578 shares of preferred stock and 3,505,800 shares of preferred stock, respectively, were undesignated and were able to be issued with such rights and powers as the Board of Directors may designate.
Series B Preferred Stock outstanding as of December 31, 2020 and 2019 consisted of 37,500 shares issued in a May 1991 private placement. Each share of Series B Preferred Stock is convertible into approximately 0.000030 shares of common stock at an effective conversion price of $22,083.75 per share of common stock, which is subject to adjustment under certain circumstances. As of December 31, 2020 and December 31, 2019, the shares of Series B Preferred Stock outstanding are convertible into 1 share of common stock. RespireRx may redeem the Series B Preferred Stock for $25,001, equivalent to $0.6667 per share, an amount equal to its liquidation preference, at any time upon 30 days prior notice.
Common Stock
There are 71,271,095 shares of the Company’s Common Stock outstanding as of December 31, 2020. After reserving for conversions of convertible debt as well as common stock purchase options and warrants exercises before accounting for incremental contract excess reserves, there were 1,870,650,077 shares of the Company’s Common Stock available for future issuances as of December 31, 2020. After accounting for incremental excess reserves required by the FirstFire Note, the EMA Note, the Crown Bridge unpaid accrued interest, and the White Lion Note as well the FirstFire Warrant, the EMA warrant, aggregating 68,777,142 and other outstanding convertible notes and all outstanding options and warrants, there were 1,801,872,935, shares of common stock available for future issuances as of December 31, 2020. Each conversion or exercise on a convertible note, or option or warrant, as appropriate reduces the excess reserve requirements. The FirstFire Note and EMA Note were converted in full in 2021 and the White Lion Note was converted in part in 2021. No warrants or options were exercised after December 31, 2020. See Note 10. Subsequent Events in the notes to our consolidated financial statements as of December 31, 2020.
| F-27 |
Common Stock Warrants
As part of our prior debt financings with FirstFire, EMA and Crown Bridge, the Company issued warrants that contained anti-dilution provisions that required a reduction to the exercise price and an increase to the number of warrant shares in the event that we issued equity instruments with a lower price than the exercise price. During the year ended December 31, 2020, we adjusted downward the warrant exercise price for these warrants. The resulting fair value of the warrants with the new exercise price was $1,440,214, recorded as a deemed dividend in our consolidated statements of stockholders’ deficiency. As the Company has an accumulated deficit, the deemed dividend was recorded within additional paid-in capital.
On September 30, 2020, the Company issued warrants exercisable into 25,377,426 of commons stock at $0.07 per share and expiring on September 30, 2023 which issuance occurred upon the conversion of all Series H Preferred Stock into common stock and warrants on September 30, 2020.
On July 30, 2020, the Company issued warrants exercisable into 375,000 shares of common stock at an exercise price of $0.07 per share and expiring on September 30, 2023.
On July 2, 2020, the Company issued warrants exercisable into 687,500 shares of common stock at an exercise price of $0.07 per share and expiring on September 30, 2023.
During the fiscal year ended December 31, 2020, inclusive of anti-dilution provisions, the Company issued warrants exercisable into 13,145,114 shares of common stock at exercise prices ranging from $0.01485 to $0.03416 of which 10,969,352 were exercised.
A summary of warrant activity for the year ended December 31, 2020 is presented below.
Number of Shares | Weighted Average Exercise Price | Weighted Average Remaining Contractual Life (in Years) | ||||||||||
| Warrants outstanding at December 31, 2019 | 219,104 | $ | 18.7109 | 3.44 | ||||||||
| Issued | 39,585,040 | 0.0521 | 2.89 | |||||||||
| Expired | (25,434 | ) | 2.9899 | |||||||||
| Exercised | (10,969,352 | ) | 0.0161 | |||||||||
| Warrants outstanding at December 31, 2020 | 28,809,352 | $ | 0.1528 | 2.64 | ||||||||
| Warrants exercisable at December 31, 2019 | 219,104 | $ | 18.7109 | 3.44 | ||||||||
| Warrants exercisable at December 31, 2020 | 28,809,352 | $ | 0.1528 | 2.64 | ||||||||
| F-28 |
The exercise prices of common stock warrants outstanding and exercisable are as follows at December 31, 2020:
| Exercise Price | Warrants
Outstanding (Shares) | Warrants
Exercisable (Shares) | Expiration Date | |||||||||
| $ | 0.0160 | 2,212,500 | 2,212,500 | May 17, 2022 | ||||||||
| $ | 0.0700 | 26,439,926 | 26,439,926 | September 30, 2023 | ||||||||
| $ | 15.000 | 19,000 | 19,000 | December 30, 2023 | ||||||||
| $ | 15.750 | 23,881 | 23,881 | April 30, 2023 | ||||||||
| $ | 27.500 | 800 | 800 | December 31, 2021 | ||||||||
| $ | 11.000 | 104,650 | 104,650 | September 29, 2022 | ||||||||
| $ | 79.300 | 8,595 | 8,595 | February 28, 2021 | ||||||||
| 28,809,352 | 29,809,352 | |||||||||||
Based on a fair value of $0.029 per share on December 31, 2020, there were 2,212,500 exercisable in-the money common stock warrants with an intrinsic value of $28,763 as of December 31, 2020.
A summary of warrant activity for the year ended December 31, 2019 is presented below.
Number of Shares | Weighted Average Exercise Price | Weighted Average Remaining Contractual Life (in Years) | ||||||||||
| Warrants outstanding at December 31, 2018 | 178,322 | $ | 22.0393 | 3.06 | ||||||||
| Issued | 47,737 | 7.9079 | 4.36 | |||||||||
| Expired | (6,955 | ) | 29.8989 | - | ||||||||
| Warrants outstanding at December 31, 2019 | 219,104 | $ | 18.7109 | 3.44 | ||||||||
| Warrants exercisable at December 31, 2018 | 178,322 | $ | 22.0393 | 3.06 | ||||||||
| Warrants exercisable at December 31, 2019 | 219,104 | $ | 18.7109 | 3.44 | ||||||||
Stock Options
On March 18, 2014, RespireRx adopted its 2014 Equity, Equity-Linked and Equity Derivative Incentive Plan (the “2014 Plan”). The Plan permits the grant of options and restricted stock with respect to up to 325,025 shares of common stock, in addition to stock appreciation rights and phantom stock, to directors, officers, employees, consultants and other service providers of the Company.
On June 30, 2015, the Board of Directors adopted the 2015 Stock and Stock Option Plan (as amended, the “2015 Plan”). As of March 31, 2020, there were 8,985,260 shares that may be issued under the 2015 Plan. On May 5, 2020 the Board of Directors increased the number of shares that may be issued under the 2015 Plan to 58,985,260. On July 31, 2020 the Board of Directors increased the number of shares that may be issued under the 2015 Plan to 158,985,260. The Company has not and does not intend to present the 2015 Plan to stockholders for approval.
Other than the change in the number of shares available under the 2015 Plan, no other changes were made to the 2015 Plan by these amendments noted above.
| F-29 |
There were no stock grants and there were stock option grants for 6,750,000 shares of RespireRx’s Common Stock during the fiscal year ended December 31, 2020 and there were no stock grants or stock option grants in the during the fiscal year ended December 31, 2019.
Information with respect to the Black-Scholes variables used in connection with the evaluation of the fair value of stock-based compensation costs and fees is provided at Note 3 Summary of Significant Accounting Policies.
A summary of stock option activity for the fiscal year ended December 31, 2020 is presented below.
Number of Shares | Weighted Average Exercise Price | Weighted Average Remaining Contractual Life (in Years) | ||||||||||
| Options outstanding at December 31, 2019 | 428,761 | $ | 33.798 | 4.98 | ||||||||
| Granted | 6,750,000 | 0.0660 | 4.63 | |||||||||
| Expired | (13,546 | ) | (65.9190 | ) | - | |||||||
| Options outstanding at December 31, 2020 | 7,165,215 | $ | 1.961 | 4.60 | ||||||||
| Options exercisable at December 31, 2020 | 6,290,215 | $ | 2.225 | 4.59 | ||||||||
The exercise prices of common stock options outstanding and exercisable were as follows at December 31, 2020:
| Exercise Price | Options Outstanding (Shares) | Options Exercisable (Shares) | Expiration Date | |||||||||
| $ | 0.072 | 5,050,000 | 4,525,000 | September 30, 2025 | ||||||||
| $ | 0.054 | 1,700,000 | 1,350,000 | July 31, 2025 | ||||||||
| $ | 7.000 | 2,168 | 2,168 | November 21, 2023 | ||||||||
| $ | 11.200 | 31,038 | 31,038 | April 5, 2023 | ||||||||
| $ | 12.500 | 1,676 | 1,676 | December 7, 2022 | ||||||||
| $ | 13.500 | 3,400 | 3,400 | July 28, 2022 | ||||||||
| $ | 14.500 | 184,942 | 184,942 | December 9, 2027 | ||||||||
| $ | 14.500 | 10,000 | 10,000 | December 9, 2027 | ||||||||
| $ | 20.000 | 28,500 | 28,500 | June 30, 2022 | ||||||||
| $ | 20.000 | 2,500 | 2,500 | July 26, 2022 | ||||||||
| $ | 39.000 | 39,500 | 39,500 | January 17, 2022 | ||||||||
| $ | 45.000 | 722 | 722 | September 2, 2021 | ||||||||
| $ | 57.500 | 261 | 261 | September 12, 2021 | ||||||||
| $ | 64.025 | 12,923 | 12,923 | August 18, 2022 | ||||||||
| $ | 64.025 | 26,179 | 26,179 | August 18, 2025 | ||||||||
| $ | 73.775 | 52,308 | 52,308 | March 31, 2021 | ||||||||
| $ | 81.250 | 16,923 | 16,923 | June 30, 2022 | ||||||||
| $ | 139.750 | 339 | 339 | March 14, 2024 | ||||||||
| $ | 159.250 | 246 | 246 | February 28, 2024 | ||||||||
| $ | 195.000 | 949 | 949 | July 17, 2022 | ||||||||
| $ | 195.000 | 641 | 641 | August 10, 2022 | ||||||||
| 7,165,215 | 6,290,215 | |||||||||||
Based on a fair value of $0.029 per share on December 31, 2020, there were no exercisable in-the-money common stock options as of December 31, 2020.
| F-30 |
A summary of stock option activity for the year ended December 31, 2019 is presented below.
Number of Shares | Weighted Average Exercise Price | Weighted Average Remaining Contractual Life (in Years) | ||||||||||
| Options outstanding at December 31, 2018 | 434,499 | $ | 35.414 | 5.90 | ||||||||
| Expired | (5,738 | ) | 156.139 | - | ||||||||
| Options outstanding at December 31, 2019 | 428,761 | $ | 33.798 | 4.98 | ||||||||
| Options exercisable at December 31, 2018 | 434,499 | $ | 35.414 | 5.90 | ||||||||
| Options exercisable at December 31, 2019 | 428,761 | $ | 33.789 | 4.98 | ||||||||
The exercise prices of common stock options outstanding and exercisable were as follows at December 31, 2019:
| Exercise Price | Options Outstanding (Shares) | Options Exercisable (Shares) | Expiration Date | |||||||||
| $ | 7.000 | 2,168 | 2,168 | November 21, 2023 | ||||||||
| $ | 11.200 | 31,038 | 31,038 | April 5, 2023 | ||||||||
| $ | 12.500 | 1,676 | 1,676 | December 7, 2022 | ||||||||
| $ | 13.500 | 3,400 | 3,400 | July 28, 2022 | ||||||||
| $ | 14.500 | 184,942 | 184,942 | December 9, 2027 | ||||||||
| $ | 14.500 | 10,000 | 10,000 | December 9, 2027 | ||||||||
| $ | 20.000 | 28,500 | 28,500 | June 30, 2022 | ||||||||
| $ | 20.000 | 2,500 | 2,500 | July 26, 2022 | ||||||||
| $ | 39.500 | 39,500 | 39,500 | January 17, 2022 | ||||||||
| $ | 45.000 | 722 | 722 | September 2, 2021 | ||||||||
| $ | 56.875 | 8,969 | 8,969 | June 30, 2020 | ||||||||
| $ | 57.500 | 261 | 261 | September 12, 2021 | ||||||||
| $ | 64.025 | 2,769 | 2,769 | August 18, 2020 | ||||||||
| $ | 64.025 | 12,923 | 12,923 | August 18, 2022 | ||||||||
| $ | 64.025 | 26,179 | 26,179 | August 18, 2025 | ||||||||
| $ | 68.250 | 879 | 879 | December 11, 2020 | ||||||||
| $ | 73.775 | 52,308 | 52,308 | March 31, 2021 | ||||||||
| $ | 81.250 | 16,923 | 16,923 | June 30, 2022 | ||||||||
| $ | 139.750 | 339 | 339 | March 14, 2024 | ||||||||
| $ | 154.700 | 775 | 755 | April 8, 2020 | ||||||||
| $ | 159.250 | 246 | 246 | February 28, 2024 | ||||||||
| $ | 166.400 | 154 | 154 | January 29, 2020 | ||||||||
| $ | 195.000 | 949 | 949 | July 17, 2022 | ||||||||
| $ | 195.000 | 641 | 641 | August 10, 2022 | ||||||||
| 428,761 | 428,761 | |||||||||||
| F-31 |
Based on a fair value of $1.00 per share on December 31, 2019, there were 0 exercisable in-the-money common stock options as of December 31, 2019.
For the years ended December 31, 2020 and 2019, stock-based compensation costs and fees included in the consolidated statements of operations consisted of general and administrative expenses of $345,500 and $0 respectively, and research and development expenses of $38,750 and $0, respectively.
Pier Contingent Stock Consideration
In connection with the merger transaction with Pier effective August 10, 2012, RespireRx issued 17,975 newly issued shares of its common stock with an aggregate fair value of $3,271,402 ($182.00 per share), based upon the closing price of RespireRx’s common stock on August 10, 2012. The shares of common stock were distributed to stockholders, convertible note holders, warrant holders, option holders, and certain employees and vendors of Pier in satisfaction of their interests and claims. The common stock issued by RespireRx represented approximately 41% of the 44,321 common shares outstanding immediately following the closing of the transaction.
The Company concluded that the issuance of any of the contingent shares to the Pier Stock Recipients was remote, as a result of the large spread between the exercise prices of these stock options and warrants as compared to the common stock trading range, the subsequent expiration or forfeiture of most of the options and warrants, the Company’s distressed financial condition and capital requirements, and that these stock options and warrants have remained significantly out-of-the-money through December 31, 2020. Accordingly, the Company considered the fair value of the contingent consideration to be immaterial and therefore did not ascribe any value to such contingent consideration. If any such shares are ultimately issued to the former Pier stockholders, the Company will recognize the fair value of such shares as a charge to operations at that time.
Reserved and Unreserved Shares of Common Stock
On January 17, 2017, the Board of Directors of the Company approved the adoption of an amendment of the Amended and Restated RespireRx Pharmaceuticals, Inc. 2015 Stock and Stock Option Plan (as amended, the “2015 Plan”). That amendment increased the shares issuable under the plan by 150,000, from 153,846 to 303,846. On December 9, and December 28, 2018, the Board of Directors further amended the 2015 Plan to increase the number of shares that may be issued under the 2015 Plan to 698,526 and 898,526 shares of the Company’s common stock. On May 5, 2020 and July 31, 2020, the Board of Directors further amended the 2015 Plan to increase the number of shares that may be issued under the 2015 Plan by 5,000,000 and 10,000,000 shares respectively. As of December 31, 2020, there are 8,704,251 shares of common stock available for issuance under the 2015 Plan.
| F-32 |
Other than the change in the number of shares available under the 2015 Plan, no other changes were made to the 2015 Plan by these amendments noted above.
At December 31, 2020, the Company had 2,000,000,000 shares of common stock authorized and 71,271,095 shares of common stock issued and outstanding. The Company has reserved 1 shares of common stock for the conversion of the Series B Preferred Stock. The Company has reserved an aggregate of 13,333,036 for the calculated amount of shares of common stock into which convertible notes may convert and an additional 53,464,642 shares of common stock for contractual reserves with respect to such notes. In addition, The Company has reserved 7,165,215 and 28,809,352 shares of the Company’s common stock for exercises of common stock purchase options granted and warrants issued respectively and an additional 15,312,500 shares of common stock for contractual reserves associated with certain of the warrants. The Company has reserved 649 shares of common stock with respect to the Pier contingent shares. There are 8,770,576 shares reserved for future issuances under the Company’s 2014 Plan and 2015 Plan. Accordingly, after taking into consideration the shares of common stock reserved for all conversions, exercises, contingent share issuances and contractual reserves, there were 1,801,872,935 shares of the Company’s common stock available for future issuances as of December 31, 2020.
7. Income Taxes
Deferred income taxes reflect the net tax effects of temporary differences between the carrying amounts of assets and liabilities for financial reporting purposes and the amounts used for income tax purposes. Significant components of the Company’s deferred tax assets as of December 31, 2020 and 2019 are summarized below.
| December 31, | ||||||||
| 2020 | 2019 | |||||||
| Capitalized research and development costs | $ | - | $ | - | ||||
| Research and development credits | 3,017,000 | 3,017,000 | ||||||
| Stock-based compensation | 3,975,000 | 3,787,000 | ||||||
| Stock options issued in connection with the payment of debt | 202,000 | 202,000 | ||||||
| Net operating loss carryforwards | 20,536,000 | 19,982,000 | ||||||
| Accrued compensation | 155,000 | 586,000 | ||||||
| Accrued interest due to related party | 146,000 | 217,000 | ||||||
| Other, net | 8,000 | 8,000 | ||||||
| Total deferred tax assets | 28,039,000 | 27,799,000 | ||||||
| Valuation allowance | (28,039,000 | ) | (27,799,000 | ) | ||||
| Net deferred tax assets | $ | - | $ | - | ||||
In assessing the potential realization of deferred tax assets, management considers whether it is more likely than not that some portion or all of the deferred tax assets will be realized. The ultimate realization of deferred tax assets is dependent upon the Company attaining future taxable income during the periods in which those temporary differences become deductible. As of December 31, 2020 and 2019, management was unable to determine that it was more likely than not that the Company’s deferred tax assets will be realized, and has therefore recorded an appropriate valuation allowance against deferred tax assets at such dates.
No federal tax provision has been provided for the years ended December 31, 2020 and 2019 due to the losses incurred during such periods. Reconciled below is the difference between the income tax rate computed by applying the U.S. federal statutory rate and the effective tax rate for the years ended December 31, 2020 and 2019.
| F-33 |
| Years Ended December 31, | ||||||||
| 2020 | 2019 | |||||||
| U. S. federal statutory tax rate | (21.0 | )% | (21.0 | )% | ||||
| Change in valuation allowance | (1.0 | )% | (1.0 | )% | ||||
| Adjustment to deferred tax asset | 22.0 | % | 22.0 | % | ||||
| Other | - | % | - | % | ||||
| Effective tax rate | 0.0 | % | 0.0 | % | ||||
As of December 31, 2020, the Company had federal and state tax net operating loss carryforwards of approximately $104,166,000 and $44,252,000, respectively. The state tax net operating loss carryforward consists of $19,673,000 for California purposes and $24,579,000 for New Jersey purposes. The difference between the federal and state tax loss carryforwards was primarily attributable to the capitalization of research and development expenses for California franchise tax purposes. The federal net operating loss carryforwards will expire at various dates from 2021 through 2040. State net operating losses expire at various dates from 2021 through 2029 for California and through 2040 for New Jersey. The Company also had federal and California research and development tax credit carryforwards that totaled approximately $1,871,000 and $1,146,000, respectively at December 31, 2020. The federal research and development tax credit carryforwards will expire at various dates from 2021 through 2032. The California research and development tax credit carryforward does not expire and will carryforward indefinitely until utilized.
While the Company has not performed a formal analysis of the availability of its net operating loss carryforwards under Internal Revenue Code Sections 382 and 383, management expects that the Company’s ability to use its net operating loss carryforwards will be limited in future periods.
The Company has not filed its federal and state tax returns for the year ended December 31, 2020, for which the Company has filed a request for extension of time to file such returns. The Company does not expect there to be any material non-filing penalties. The Company intends to file such returns as soon as practical.
8. Related Party Transactions
Dr. Arnold S. Lippa and Jeff E. Margolis, officers and directors of the Company since March 22, 2013, have indirect ownership interests and managing memberships in Aurora Capital LLC (“Aurora”) through interests held in its members, and Jeff. E. Margolis is also an officer of Aurora. Aurora is a boutique investment banking firm specializing in the life sciences sector that limits its securities related activities primarily to investment banking services.
A description of advances and notes payable to officers is provided at Note 4. Notes Payable – Advances from and Notes Payable to Officer.
9. Commitments and Contingencies
Pending or Threatened Legal Action and Claims
The Company is periodically the subject of various pending and threatened legal actions and claims. In the opinion of management of the Company, adequate provision has been made in the Company’s consolidated financial statements as of December 31, 2020 and 2019 with respect to such matters. See Note 5. Settlement and Payment Agreements to the consolidated financial statements as of December 31, 2020 for additional items and details.
| F-34 |
Significant Agreements and Contracts
Consulting Agreements
Richard Purcell, the Company’s Senior Vice President of Research and Development since October 15, 2014, provides his services to the Company on a month-to-month basis through his consulting firm, DNA Healthlink, Inc., through which the Company has contracted for his services, for a monthly cash fee of $12,500. Cash compensation expense pursuant to this agreement totaled $112,500 and $150,000 for the fiscal years ended December 31, 2020 and 2019, respectively, which is included in research and development expenses in the Company’s consolidated statements of operations for such periods.
The Company entered into a consulting contract with David Dickason effective September 15, 2020 pursuant to which Mr. Dickason was appointed to and serves as the Company’s Senior Vice President of Pre-Clinical Product Development on an at-will basis at the rate of $250 per hour.
Employment Agreements
Effective on May 6, 2020, Timothy Jones was appointed as RespireRx’s President and Chief Executive Officer and entered into an employment agreement as of that date. In addition, Mr. Jones has continued to serve as a member of the Company’s Board of Directors, a position he has held since January 28, 2020. On November 19, 2019, Mr. Jones became an advisor to the Company’s Board of Directors, a position he held until January 27, 2020. Under the employment agreement, a provisional period of “at will” employment expired on July 31, 2020. Neither party terminated the employment agreement prior to July 31, 2020, and on that date all rights and obligations under the agreement were deemed effective, including with respect to the certain economic obligations of the Company upon termination of Mr. Jones’ employment. The Board of Directors and Mr. Jones agreed to continue the employment agreement after the initial provisional period. The employment agreement has a termination date of September 30, 2023 and will automatically extend annually, upon the same terms and conditions, for successive periods of one year, unless either party provides written notice of its intention not to extend the term of the agreement at least 90 days prior to the applicable renewal date. On July 31, 2020, the employment agreement was amended. The terms of the amended agreement call for a base salary through September 30, 2020 of $300,000 per year which may remain accrued but unpaid at the discretion of the Board of Directors until such time as at least $2,500,000 has been raised. As of December 31, 2020, Mr. Jones base salary remained $300,000 per year. If $10,000,000 or more has been raised by September 30, 2021, Mr. Jones’ base salary would be increased to $375,000 per year. Otherwise, it would remain at $300,000 annually unless increased pursuant to the employment agreement or by the Board of Directors. Mr. Jones’ base salary is subject to cost of living increases. Mr. Jones was eligible for a guaranteed bonus of $200,000 on October 31,2020. Mr. Jones is also entitled to bonuses of $200,000 on March 31, 2021 and $150,000 each six months thereafter on each March 31st and September 30th thereafter, unless the agreement is earlier terminated. The guaranteed bonus of $200,000 that was due on October 31, 2020 was not paid and is accrued and payable as of that date. At the end of the provisional period, pursuant to the employment agreement, Mr. Jones was granted an option grant for the purchase of 1,000,000 shares of the Company’s common stock upon the expiration of the provisional period which has since been adjusted to 100,000 shares after the reverse stock-split and is presented in the options tables in our consolidated financial statements as of December 31, 2020 on a post reverse stock-split basis. In addition, until such time as the Company establishes comparable benefits, Mr. Jones is entitled to $1,200 per month on a tax equalized basis for health insurance and $1,000 per month on a tax equalized basis for term life insurance plus a disability policy. Mr. Jones is entitled to be reimbursed for business expenses. Mr. Jones would be entitled to a $12,000 tax equalized annual automobile allowance after the Company has raised $10,000,000. In addition, on July 31, 2020, the Board of Directors granted Mr. Jones a discretionary bonus that was a grant of an option to purchase 16,000,000 shares of common stock expiring on July 31, 2025 at an exercise price equal to the closing price of the Company’s common stock on July 31, 2020 of $0.0072 which has been adjusted to 1,600,000 shares and an exercise price of $0.072 after the reverse stock-split, and which is presented in the options tables in our consolidated financial statements as of December 31, 2020 on a post reverse stock-split basis, 25% of which vested immediately, 25% of which vested on September 30, 2020, December 31, 2020 and March 31, 2021. Upon commencement of Mr. Jones’ employment agreement on May 6, 2020, Mr. Jones was no longer eligible to receive fees for his participation as a member of the Board of Directors. For the fiscal year ended December 31, 2020, the Company accrued $436,059 of compensation and related benefits for Mr. Jones in the form of Board of Directors advisory fees, salary and benefits and bonus, of which Mr. was paid $16,073 in cash and exchanged $28,218 of the accrued obligation for Series H Preferred Stock as discussed below. These amounts are included in accounts payable and accrued expenses and in accrued compensation in the Company’s consolidated balance sheet as of December 31, 2020. On September 30, 2020, Mr. Jones, pursuant to an exchange agreement, forgave $28,218 of accrued Board of Directors and other fees owed to him in exchange for 28.218 shares of Series H Preferred Stock which, on the same day, was converted into 4,409,063 shares of Common Stock and a warrant to purchase 4,409,063 shares of Common Stock, both of which were adjusted to 440,906 shares of Common Stock and warrants respectively after the reverse stock-split and which are presented on a reverse stock-split basis in our consolidated financial statements as of December 31, 2020.
Effective May 6, 2020, with the appointment of Timothy Jones as RespireRx’s President and Chief Executive Officer, Dr. Lippa resigned the interim officer positions of Interim Chief Executive Officer and Interim President, positions that Dr. Lippa had assumed on October 12, 2018 after the resignation of Dr. James Manuso on September 30, 2018. Dr. Lippa continues to serve as RespireRx’s Executive Chairman and as a member of the Board of Directors as well as the Company’s Chief Scientific Officer. Dr. Lippa has continued to serve as the Company’s Executive Chairman and as a member of the Board of Directors. On August 18, 2015, Dr. Lippa was named Chief Scientific Officer of the Company, and the Company entered into an employment agreement with Dr. Lippa in that capacity. Pursuant to the agreement, which was for an initial term through September 30, 2018 (and which automatically extended on September 30, 2018, 2019 and 2020 and will automatically extend annually, upon the same terms and conditions, for successive periods of one year, unless either party provides written notice of its intention not to extend the term of the agreement at least 90 days prior to the applicable renewal date), Dr. Lippa earned an annual base salary of $300,000. Dr. Lippa is also eligible to earn a performance-based annual bonus award of up to 50% of his base salary, based upon the achievement of annual performance goals established by the Board of Directors in consultation with the executive prior to the start of such fiscal year, or any amount at the discretion of the Board of Directors. Additionally, Dr. Lippa has been granted stock options on several occasions and is eligible to receive additional awards under the Company’s Plans at the discretion of the Board of Directors. Dr. Lippa did not receive any option to purchase shares of common stock during fiscal year ended December 31, 2020. Dr. Lippa is also entitled to receive, until such time as the Company establishes a group health plan for its employees, $1,200 per month, on a tax-equalized basis, as additional compensation to cover the cost of health coverage and up to $1,000 per month, on a tax-equalized basis, as reimbursement for a term life insurance policy and disability insurance policy. Dr. Lippa is also entitled to be reimbursed for business expenses. Additional information with respect to the stock options granted to Dr. Lippa is provided at Note 6 to the Company’s consolidated financial statements for the fiscal years ended December 31, 2020 and 2019. Cash compensation inclusive of employee benefits accrued pursuant to this agreement totaled $339,600 for each of the fiscal years ended December 31, 2020 and 2019, respectively, which amounts are included in accrued compensation and related expenses in the Company’s consolidated balance sheet at December 31, 2020 and 2019, and in research and development expenses in the Company’s consolidated statement of operations for the fiscal years ended December 31, 2020 and 2020. Dr. Lippa does not receive any additional compensation for serving as Executive Chairman and on the Board of Directors.
| F-35 |
On March 22, 2020, July 13, 2020 and September 30, 2020, Dr. Lippa, forgave an aggregate of $853,000 of accrued compensation and benefits. On March 22, 2020, Dr, Lippa received 4,500,000 shares Common Stock for $153,000 of forgiven compensation, which shares of Common Stock were adjusted to 450,000 shares of Common Stock on a post reverse stock-split basis. On July 13, 2020, pursuant to an exchange agreement, Dr. Lippa forgave $600,000 of accrued compensation and benefits and in exchange received 600 shares of Series H Preferred Stock. On September 30, 2020, pursuant to an additional exchange agreement, Dr. Lippa forgave $100,000 of accrued compensation and benefits and in exchange received 100 shares of Series H Preferred Stock. Between July 13, 2020 and September 30, 2020, Dr. Lippa earned 2.6333333 shares of Series H Preferred Stock as dividends in-kind. On July 13, 2020 and September 30, 2020, Dr. Lippa contributed all of his Series H Preferred Stock to a family trust. On September 30, 2020, the family trust converted all of its Series H Preferred Stock into 109,786,458 shares of RespireRx Common Stock and a warrant to purchase 109,786,458 shares of Common Stock which were subsequently adjusted to 10,978,645 shares of Common Stock and warrants to purchase 10,978,645 shares of Common Stock.
On August 18, 2015, the Company also entered into an employment agreement with Jeff E. Margolis, in his role at that time as Vice President, Secretary and Treasurer. Pursuant to the agreement, which was for an initial term through September 30, 2016 and later amended (and which automatically extended on September 30, 2016, 2017, 2018 and 2019 and will automatically extend annually, upon the same terms and conditions for successive periods of one year, unless either party provides written notice of its intention not to extend the term of the agreement at least 90 days prior to the applicable renewal date), Mr. Margolis currently receives an annual base salary of $300,000, and is eligible to receive performance-based annual bonus awards based upon the achievement of annual performance goals established by the Board of Directors in consultation with the executive prior to the start of such fiscal year. Additionally, Mr. Margolis has granted stock options on several occasions and is eligible to receive additional awards under the Company’s Plans at the discretion of the Board of Directors. Mr. Margolis is also entitled to receive, until such time as the Company establishes a group health plan for its employees, $1,200 per month, on a tax-equalized basis, as additional compensation to cover the cost of health coverage and up to $1,000 per month, on a tax-equalized basis, as reimbursement for a term life insurance policy and disability insurance policy. Mr. Margolis is also entitled to be reimbursed for business expenses. Additional information with respect to the stock options granted to Mr. Margolis is provided at Note 6 to the Company’s consolidated financial statements for fiscal years ended December 31, 2020 and 2019. Recurring cash compensation accrued pursuant to this amended agreement totaled $321,600 for the fiscal year ended December 31, 2020 and 2019 which amounts are included in accrued compensation and related expenses in the Company’s consolidated balance sheet December 31, 2020 and 2019, and in general and administrative expenses in the Company’s consolidated statement of operations.
On March 22, 2020, July 13, 2020 and September 30, 2020, Mr. Margolis, forgave an aggregate of $803,000 of accrued compensation and benefits. On March 22, 2020, Mr. Margolis received 4,500,000 shares Common Stock for $153,000 of forgiven compensation, which shares of Common Stock were adjusted to 450,000 shares of Common Stock on a post reverse stock-split basis. On July 13, 2020, pursuant to an exchange agreement, Mr. Margolis forgave $500,000 of accrued compensation and benefits and in exchange received 500 shares of Series H Preferred Stock. On September 30, 2020, pursuant to an additional exchange agreement, Mr. Margolis forgave $150,000 of accrued compensation and benefits and in exchange received 150 shares of Series H Preferred Stock. Between July 13, 2020 and September 30, 2020, Mr. Margolis earned 2.194444 shares of Series H Preferred Stock as dividends in-kind. On July 13, 2020 and September 30, 2020, Mr. Margolis contributed all of his Series H Preferred Stock to three family trusts. On September 30, 2020, the family trusts converted all of their Series H Preferred Stock into 101,905,382 shares of RespireRx Common Stock and a warrant to purchase 101,905,382 shares of Common Stock which were subsequently adjusted to 10,190,538 shares of Common Stock and warrants to purchase 10,190,538 shares of Common Stock.
The employment agreements between the Company and Dr. Lippa, and Mr. Margolis (prior to the 2017 amendment), respectively, provided that the payment obligations associated with the first year base salary were to accrue, but no payments were to be made, until at least $2,000,000 of net proceeds from any offering or financing of debt or equity, or a combination thereof, was received by the Company, at which time scheduled payments were to commence. Dr. Lippa, and Mr. Margolis (who are each also directors of the Company) have each agreed, effective as of August 11, 2016, to continue to defer the payment of such amounts indefinitely, until such time as the Board of Directors of the Company determines that sufficient capital has been raised by the Company or is otherwise available to fund the Company’s operations on an ongoing basis.
UWMRF Patent License Agreement
On August 1, 2020, RespireRx exercised its option pursuant to its option agreement dated March 2, 2020, between RespireRx and UWM Research Foundation, an affiliate of the University of Wisconsin-Milwaukee (“UWMRF”). Upon exercise RespireRx and UWMRF executed the UWMRF Patent License Agreement effective August 1, 2020 pursuant to which RespireRx licensed the identified intellectual property.
Under the UWMRF Patent License Agreement, the Company has an exclusive license to commercialize GABAkine products based on UWMRF’s rights in certain patents and patent applications, and a non-exclusive license to commercialize products based on UWMRF’s rights in certain technology that is not the subject of the patents or patent applications. UWMRF maintains the right to use, and, upon the approval of the Company, to license, these patent and technology rights for any non-commercial purpose, including research and education. The UWMRF Patent License Agreement expires upon the later of the expiration of the Company’s payment obligations to UWMRF or the expiration of the last remaining licensed patent granted thereunder, subject to early termination upon the occurrence of certain events. The License Agreement also contains a standard indemnification provision in favor of UWMRF and confidentiality provisions obligating both parties.
| F-36 |
Under the UWMRF Patent License Agreement, in consideration for the licenses granted, the Company will pay to UWMRF the following: (i) patent filing and prosecution costs incurred by UWMRF prior to the effective date, paid in yearly installments over three years from the Effective Date; (ii) annual maintenance fees, beginning on the second anniversary of the Effective Date, which annual maintenance fees terminate upon the Company’s payment of royalties pursuant to clause (iv) below; (iii) milestone payments, paid upon the occurrence of certain dosing events of patients during clinical trials and certain approvals by the FDA; and (iv) royalties on net sales of products developed with the licenses, subject to minimum annual payments and to royalty rate adjustments based on whether separate royalty payments by the Company yield an aggregate rate beyond a stated threshold. The Company has also granted UWMRF certain stock appreciation rights with respect to the Company’s neuromodulator programs, subject to certain limitations, and will pay to UWMRF certain percentages of revenues generated from sublicenses of the licenses provided under the UWMRF Patent License Agreement by the Company to third parties.
University of Illinois 2014 Exclusive License Agreement
On June 27, 2014, the Company entered into an Exclusive License Agreement (the “2014 License Agreement”) with the University of Illinois, the material terms of which were similar to a License Agreement between the parties that had been previously terminated on March 21, 2013. The 2014 License Agreement became effective on September 18, 2014, upon the completion of certain conditions set forth in the 2014 License Agreement, including: (i) the payment by the Company of a $25,000 licensing fee, (ii) the payment by the Company of outstanding patent costs aggregating $15,840, and (iii) the assignment to the University of Illinois of rights the Company held in certain patent applications, all of which conditions were fulfilled.
The 2014 License Agreement granted the Company (i) exclusive rights to several issued and pending patents in numerous jurisdictions and (ii) the non-exclusive right to certain technical information that is generated by the University of Illinois in connection with certain clinical trials as specified in the 2014 License Agreement, all of which relate to the use of cannabinoids for the treatment of sleep related breathing disorders. The Company is developing dronabinol (Δ9-tetrahydrocannabinol), a cannabinoid, for the treatment of OSA, the most common form of sleep apnea.
The 2014 License Agreement provides for various commercialization and reporting requirements commencing on June 30, 2015. In addition, the 2014 License Agreement provides for various royalty payments, including a royalty on net sales of 4%, payment on sub-licensee revenues of 12.5%, and a minimum annual royalty beginning in 2015 of $100,000, which is due and payable on December 31 of each year beginning on December 31, 2015. The minimum annual royalty obligation of $100,000 due on December 31, 2020, was extended to April 19, 2021 and was paid in full on April 1, 2021. One-time milestone payments may become due based upon the achievement of certain development milestones. $350,000 will be due within five days after the dosing of the first patient is a Phase III human clinical trial anywhere in the world. $500,000 will be due within five days after the first NDA filing with FDA or a foreign equivalent. $1,000,000 will be due within twelve months of the first commercial sale. One-time royalty payments may also become due and payable. Annual royalty payments may also become due. In the year after the first application for market approval is submitted to the FDA or a foreign equivalent and until approval is obtained, the minimum annual royalty will increase to $150,000. In the year after the first market approval is obtained from the FDA or a foreign equivalent and until the first sale of a product, the minimum annual royalty will increase to $200,000. In the year after the first commercial sale of a product, the minimum annual royalty will increase to $250,000.
During the fiscal years ended December 31, 2020 and 2019, the Company recorded charges to operations of $100,000, respectively, with respect to its 2020 and 2019 minimum annual royalty obligation, which is included in research and development expenses in the Company’s consolidated statement of operations for the fiscal years ended December 31, 2020 and 2019. The Company did not pay the amount due on December 31, 2020 for which the Company was granted an extension until April 19, 2021 and which was paid in full on April 1, 2021.
| F-37 |
University of Alberta License Agreement
On May 18, 2018, the Company received a letter from counsel claiming to represent TEC Edmonton and The Governors of the University of Alberta, which purported to terminate, effective December 12, 2017, the license agreement dated May 9, 2007 (as subsequently amended) between the Company and The Governors of the University of Alberta. The Company, through its counsel, disputed any grounds for termination and notified the representative that it invoked Section 13 of that license agreement, which mandates a meeting to be attended by individuals with decision-making authority to attempt in good faith to negotiate a resolution to the dispute. In February 2019, the Company and TEC Edmonton tentatively agreed to terms acceptable to all parties to establish a new license agreement and the form of a new license agreement. However, after reaching that tentative agreement, the Company has re-evaluated that portion of its ampakine program and has decided not to enter into a new agreement at this time. The lack of entry into a new agreement at this time does not affect the Company’s other ampakine programs and permits the Company to reallocate resources to those programs, including, but not limited to ADHD, FXS, SCI and CNS-driven Disorders.
Noramco Inc. - Dronabinol Development and Supply Agreement
On September 4, 2018, RespireRx entered into a dronabinol Development and Supply Agreement with Noramco Inc., one of the world’s major dronabinol manufacturers. Under the terms of the Agreement, Noramco agreed to (i) provide all of the active pharmaceutical ingredient (“API”) estimated to be needed for the clinical development process for both the first- and second-generation products (each a “Product” and collectively, the “Products”), three validation batches for New Drug Application (“NDA”) filing(s) and adequate supply for the initial inventory stocking for the wholesale and retail channels, subject to certain limitations, (ii) maintain or file valid drug master files (“DMFs”) with the FDA or any other regulatory authority and provide the Company with access or a right of reference letter entitling the Company to make continuing reference to the DMFs during the term of the agreement in connection with any regulatory filings made with the FDA by the Company, (iii) participate on a development committee, and (iv) make available its regulatory consultants, collaborate with any regulatory consulting firms engaged by the Company and participate in all FDA or Drug Enforcement Agency (“DEA”) meetings as appropriate and as related to the API.
In consideration for these supplies and services, the Company has agreed to purchase exclusively from Noramco during the commercialization phase all API for its Products as defined in the Development and Supply Agreement at a pre-determined price subject to certain producer price adjustments and agreed to Noramco’s participation in the economic success of the commercialized Product or Products up to the earlier of the achievement of a maximum dollar amount or the expiration of a period of time.
Transactions with Biovail Laboratories International SRL
Beginning in March 2010, the Company entered into a series of asset purchase and license agreements with Biovail Laboratories International SRL later merged with Valeant Pharmaceuticals International, Inc. which was later renamed Bausch Health Companies Inc. (“Biovail”).
In March 2011, the Company entered into a new agreement with Biovail to reacquire the ampakine compounds, patents and rights that Biovail had acquired from the Company in March 2010. The new agreement provided for potential future payments of up to $15,150,000 by the Company based upon the achievement of certain developments, including new drug application submissions and approval milestones pertaining to an intravenous dosage form of the ampakine compounds for respiratory depression, a therapeutic area not currently pursued by the Company. Biovail is also eligible to receive additional payments of up to $15,000,000 from the Company based upon the Company’s net sales of an intravenous dosage form of the compounds for respiratory depression.
| F-38 |
At any time following the completion of Phase 1 clinical studies and prior to the end of Phase 2A clinical studies, Biovail retains an option to co-develop and co-market intravenous dosage forms of an ampakine compound as a treatment for respiratory depression and vaso-occlusive crises associated with sickle cell disease. In such an event, the Company would be reimbursed for certain development expenses to date and Biovail would share in all such future development costs with the Company. If Biovail makes the co-marketing election, the Company would owe no further milestone payments to Biovail and the Company would be eligible to receive a royalty on net sales of the compound by Biovail or its affiliates and licensees.
Summary of Principal Cash Obligations and Commitments
The following table sets forth the Company’s principal cash obligations and commitments for the next five fiscal years as of December 31, 2020, aggregating $2,885,270. Employment agreement amounts included in the 2021 column represent amounts contractually due from January 1, 2021 through September 30, 2021 or in one case, September 30, 2023 when such contracts expire unless extended pursuant to the terms of the contracts.
| Payments Due By Year | ||||||||||||||||||||||||
| Total | 2021 | 2022 | 2023 | 2024 | 2025 | |||||||||||||||||||
| License agreements | $ | 560,370 | $ | 100,000 | $ | 115,092 | $ | 115,093 | $ | 130,185 | $ | 100,000 | ||||||||||||
| Employment agreements (1) | 2,294,900 | 1,100,600 | 639,600 | 554,700 | - | - | ||||||||||||||||||
| Total | $ | 2,855,270 | $ | 1,200,600 | $ | 754,692 | $ | 669,793 | $ | 130,185 | $ | 100,000 | ||||||||||||
(1) The payment of certain of such amounts has been deferred indefinitely, as described above in “Employment Agreements”.
10. Subsequent Events
As of 5pm eastern time on January 5, 2021, the Company effected a ten to one (10:1) reverse stock split of its Common Stock by filing the Sixth Amendment of Second Restated Certificate of Incorporation of RespireRx Pharmaceuticals Inc. with the Secretary of State of the State of Delaware on January 4, 2021. The financial statements have been prepared as of December 31, 2020 and for the fiscal year then ended on a post-reverse stock split basis.
After a downgrade of the trading of our Common Stock to the OTC Pink Market on December 10, 2020, the reverse stock split described above occurred on January 5, 2021 and the Common Stock began trading on a post-reverse stock split basis on the OTC Markets OTC Pink Market from January 6, 2021 through Friday, February 5, 2021 while measuring for thirty calendar days, compliance with the uplisting requirements for listing on the OTC Markets OTCQB. The stock was upgraded to the OTCQB on Monday, February 8, 2021.
A purchase notice was sent to White Lion on February 19, 2021 was for 3,600,000 shares of Common Stock resulting in net proceeds after closing costs of $2,070 aggregating $115,229. The total of all shares sold pursuant to purchase notices (including three purchase notices in the fiscal year ended December 31, 2020 and the one February 19, 2021 purchase notice is 11,500,000 which is all of the shares registered and offered for sale is the registration statement on Form S-1 that became effective on October 29, 2020. There are no shares available for sale under that registration statement. In order for the Company to issue additional purchase notices to White Lion, the Company would either have to file a new registration statement or amend the current registration statement covering shares representing any remaining amounts available under the White Lion EPA, or up to $1,711,581.
| F-39 |
The FirstFire Note dated July 2, 2020 was paid in full by conversion as follows:
| Conversion Date | Principal Converted | Interest Converted | Costs paid by the Company by conversion | Conversion Price per Share | Shares of Common Stock Issued | |||||||||||||||
| January 19, 2021 | $ | 30,000 | $ | 0 | $ | 0 | $ | 0.02 | 1,500,000 | |||||||||||
| February 4, 2021 | $ | 37,500 | $ | 0 | $ | 0 | $ | 0.02 | 1,875,000 | |||||||||||
| February 16, 2021 | $ | 50,000 | $ | 0 | $ | 0 | $ | 0.02 | 2,500,000 | |||||||||||
| March 3, 2021 | $ | 20,000 | $ | 6,875 | $ | 0 | $ | 0.02 | 1,343,750 | |||||||||||
| Total | $ | 137,500 | $ | 6,875 | $ | 0 | 7,218,750 | |||||||||||||
The EMA Note dated July 30, 2020 was paid in full by conversion as follows:
| Conversion Date | Principal Converted | Interest Converted | Costs paid by the Company by conversion | Conversion Price per Share | Shares of Common Stock Issued | |||||||||||||||
| February 4, 2021 | $ | 19,000 | $ | 0 | $ | 1,000 | $ | 0.02 | 1,000,000 | |||||||||||
| February 10, 2021 | $ | 19,000 | $ | 0 | $ | 1,000 | $ | 0.02 | 1,000,000 | |||||||||||
| February 12, 2021 | $ | 19,000 | $ | 0 | $ | 1,000 | $ | 0.02 | 1,000,000 | |||||||||||
| March 3, 2021 | $ | 18,000 | $ | 4,136 | $ | 1,000 | $ | 0.02 | 1,156,807 | |||||||||||
| Total | $ | 75,000 | $ | 4,136 | $ | 4,000 | 4,156,807 | |||||||||||||
On March 15, 2021, the White Lion Commitment Note was repaid in part by conversion as follows:
| Conversion Date | Principal Converted | Interest Converted | Costs paid by the Company by conversion | Conversion Price per Share | Shares of Common Stock Issued | |||||||||||||||
| March 15, 2021 | $ | 25,000 | $ | 0 | $ | 0 | $ | 0.02 | 1,250,000 | |||||||||||
On February 17, 2021, the Company and FirstFire entered into a Securities Purchase Agreement (the “FirstFire Feb 2021 SPA”) pursuant to which FirstFire provided a sum of $100,000 (the “FirstFire Feb 2021 Consideration”) to the Company, in return for a convertible promissory note (the “FirstFire Feb 2021 Note”) with a face amount of $112,000 (which difference in value as compared to the FirstFire Feb 2021 Consideration is due to an original issue discount of $12,000) and 2,000,000 commitment shares of Common Stock, a piggy-back registration rights agreement and other agreements and obligations. The net proceeds of the First Fire Feb 2021 Consideration, which were received by the Company on February 19, 2021, equal $97,500 after payment of $2,500 in FirstFire’s legal fees. The FirstFire Feb 2021 Note bears interest at 10% per annum. The terms of the FirstFire Feb 2021 Note require that the Company reserve 18,060,000 shares of Common Stock or three times the number of shares into which the FirstFire Feb 2021 Note may convert, but no less than the initial number of shares of Common Stock that must be reserved.
On March 31, 2021, the Company and EMA entered into a Securities Purchase Agreement (the “EMA March 2021 SPA”) pursuant to which EMA provided a sum of $100,000 (the “EMA March 2021 Consideration”) to the Company, in return for a convertible promissory note (the “EMA March 2021 Note”) with a face amount of $112,500 (which difference in value as compared to the EMA March 2021 Consideration is due to an original issue discount of $12,500) and a warrant exercisable for five years at $0.02 per share on a cash or cashless basis, into 2,400,000 shares of Common Stock, a piggy-back registration rights agreement and other agreements and obligations. The net proceeds of the EMA March 2021 Consideration, which were received by the Company on April 1, 2021, equal $96,750 after payment of $2,750 in EMA’s legal fees. And $500 of EMA’s due diligence fees. The EMA March 2021 Note bears interest at 10% per annum and matures on March 31, 2022. The EMA March 2021 Note is convertible at a fixed conversion price of $0.02 per share of Common Stock. The terms of the EMA March 2021 Note require that the Company reserve the greater of (i) 26,602,500 shares of Common Stock or (ii) three times the number of shares into which the EMA March 2021 Note may convert and into which the warrant may exercise.
| F-40 |
RESPIRERX PHARMACEUTICALS INC.
AND SUBSIDIARY
CONDENSED CONSOLIDATED BALANCE SHEETS
| September 30, 2021 | December 31, 2020 | |||||||
| (unaudited) | ||||||||
| ASSETS | ||||||||
| Current assets: | ||||||||
| Cash | $ | 556 | $ | 825 | ||||
| Deferred financing costs | 167,976 | 52,609 | ||||||
| Prepaid expenses | 61,449 | 31,653 | ||||||
| Total current assets | 229,981 | 85,087 | ||||||
| Total assets | $ | 229,981 | $ | 85,087 | ||||
| LIABILITIES AND STOCKHOLDERS’ DEFICIENCY | ||||||||
| Current liabilities: | ||||||||
| Accounts payable and accrued expenses, including amounts owed to related parties | $ | 5,058,227 | $ | 4,923,947 | ||||
| Accrued compensation and related expenses | 2,408,759 | 1,540,809 | ||||||
| Convertible notes payable, including accrued interest of $121,493 and $85,693 at September 30, 2021 and December 31, 2020, respectively, which includes accrued interest to related parties (Note 4) | 559,790 | 414,860 | ||||||
| Note payable to SY Corporation, including accrued interest of $447,266 and $411,385 at September 30, 2021 and December 31, 2020, respectively (payment obligation currently in default – Note 4) | 830,202 | 864,551 | ||||||
| Notes payable to officer, including accrued interest (Note 4) | 229,390 | 213,067 | ||||||
| Notes payable to former officer, including accrued interest (Note 4) | 200,519 | 186,565 | ||||||
| Other short-term notes payable | 35,982 | 4,608 | ||||||
| Total current liabilities | 9,322,869 | 8,148,407 | ||||||
| Long-term liabilities | ||||||||
| Long-term accounts payable associated with payment settlement agreements (Note 5) | 332,000 | - | ||||||
| Total long-term liabilities | 332,000 | - | ||||||
| Total liabilities | 9,654,869 | - | ||||||
| Commitments and contingencies (Note 8) | ||||||||
| Stockholders’ deficiency: (Note 6) | ||||||||
| Series B convertible preferred stock, $0.001 par value; $0.6667 per share liquidation preference; aggregate liquidation preference $25,001; shares authorized, issued and outstanding: 37,500; common share issuable upon conversion at 0.000030 common shares per Series B share: 1 | 21,703 | 21,703 | ||||||
| Common stock, $0.001 par value; shares authorized: 2,000,000,000; shares issued and outstanding: 90,396,596 at September 30, 2021 and 71,271,095 at December 31, 2020, respectively (Note 2 and Note 6) | 90,397 | 71,271 | ||||||
| Additional paid-in capital | 163,644,453 | 162,654,002 | ||||||
| Accumulated deficit | (173,181,441 | ) | (170,810,296 | ) | ||||
| Total stockholders’ deficiency | (9,424,888 | ) | (8,063,320 | ) | ||||
| Total liabilities and stockholders’ deficiency | $ | 229,981 | $ | 85,087 | ||||
See accompanying notes to condensed consolidated financial statements (unaudited).
| F-41 |
RESPIRERX PHARMACEUTICALS INC.
AND SUBSIDIARY
CONDENSED CONSOLIDATED STATEMENTS OF OPERATIONS
(Unaudited)
| Three-Months Ended | Nine-Months Ended | |||||||||||||||
| September 30, | September 30, | |||||||||||||||
| 2021 | 2020 | 2021 | 2020 | |||||||||||||
| Operating expenses: | ||||||||||||||||
| General and administrative, including related parties | $ | 423,558 | $ | 1,140,204 | $ | 1,495,103 | $ | 1,969,223 | ||||||||
| Research and development, including related parties | 157,751 | 171,776 | 550,343 | 480,242 | ||||||||||||
| Total operating expenses | 581,309 | 1,311,980 | 2,045,446 | 2,449,465 | ||||||||||||
| Loss from operations | (581,309 | ) | (1,311,980 | ) | (2,045,446 | ) | (2,449,465 | ) | ||||||||
| Gain on warrant exchange | - | - | 1,099 | - | ||||||||||||
| Gain/(Loss) on extinguishment or settlement of debt and other liabilities | 62,548 | (65,906 | ) | 62,548 | (389,902 | ) | ||||||||||
| Interest expense, including related parties | (228,253 | ) | (78,678 | ) | (459,566 | ) | (409,994 | ) | ||||||||
| Foreign currency transaction gain (loss) | 38,332 | (22,791 | ) | 70,220 | 7,151 | |||||||||||
| Net loss | $ | (708,682 | ) | $ | (1,479,355 | ) | $ | (2,371,145 | ) | $ | (3,242,210 | ) | ||||
| Deemed dividend associated with most favored nation provisions of convertible notes | $ | (378,042 | ) | $ | - | $ | (378,042 | ) | $ | - | ||||||
| Net loss attributable to common stockholders | $ | (1,086,724 | ) | $ | 1,479,355 | $ | (2,749,187 | ) | $ | - | ||||||
| Net loss per common share - basic and diluted | $ | (0.01 | ) | $ | (0.01 | ) | $ | (0.03 | ) | $ | (0.02 | ) | ||||
| Weighted average common shares outstanding - basic and diluted | 90,396,596 | 22,435,203 | 83,757,619 | 13,179,304 | ||||||||||||
See accompanying notes to condensed consolidated financial statements (unaudited).
| F-42 |
RESPIRERX PHARMACEUTICALS INC.
AND SUBSIDIARY
CONDENSED CONSOLIDATED STATEMENTS OF STOCKHOLDERS’ DEFICIENCY
(Unaudited)
Nine-months and Three-months Ended September 30, 2021
| Series B | ||||||||||||||||||||||||||||
| Convertible | ||||||||||||||||||||||||||||
| Preferred Stock | Common Stock | Additional | Total | |||||||||||||||||||||||||
| Par | Paid-in | Accumulated | Stockholders’ | |||||||||||||||||||||||||
| Shares | Amount | Shares | Value | Capital | Deficit | Deficiency | ||||||||||||||||||||||
| Balance, December 31, 2020 | 37,500 | $ | 21,703 | 71,271,095 | $ | 71,271 | $ | 162,654,002 | $ | (170,810,296 | ) | $ | (8,063,320 | ) | ||||||||||||||
| Sale of common stock | - | - | 3,600,000 | 3,600 | 113,699 | - | 117,299 | |||||||||||||||||||||
| Costs of stock issuance | - | - | - | - | (52,609 | ) | - | (52,609 | ) | |||||||||||||||||||
| Issuance of note commitment shares and beneficial conversion feature | - | - | 2,000,000 | 2,000 | 95,500 | - | 97,500 | |||||||||||||||||||||
| Issuance of common stock upon conversion of convertible notes | - | - | 12,625,557 | 12,626 | 239,885 | - | 252,511 | |||||||||||||||||||||
| Stock -based compensation | - | - | - | - | 44,250 | - | 44,250 | |||||||||||||||||||||
| Adjustment due to reverse stock split | - | - | (56 | ) | - | - | - | - | ||||||||||||||||||||
| Net loss | - | - | - | - | - | (850,249 | ) | (850,249 | ) | |||||||||||||||||||
| Balance, March 31, 2021 | 37,500 | $ | 21,703 | 89,496,596 | $ | 89,497 | $ | 163,094,727 | $ | (171,660,545 | ) | $ | (8,454,618 | ) | ||||||||||||||
| Issuance of common stock upon cashless warrant exercise | - | - | 900,000 | 900 | (900 | ) | - | - | ||||||||||||||||||||
| Stock-based compensation | - | - | - | - | 7,500 | - | 7,500 | |||||||||||||||||||||
| Gain on warrant exchanges | - | - | - | - | (1,099 | ) | - | (1,099 | ) | |||||||||||||||||||
| Note discounts | - | - | - | - | 443,550 | - | 443,550 | |||||||||||||||||||||
| Net loss | - | - | - | - | - | (812,214 | ) | (812,214 | ) | |||||||||||||||||||
| Balance,June 30, 2021 | 37,500 | $ | 21,703 | 90,396,596 | $ | 90,397 | $ | 163,543,778 | $ | (172,472,759 | ) | $ | (8,816,881 | ) | ||||||||||||||
| Issuance of note with beneficial conversion features, other note discounts and warrant | - | - | - | - | 98,175 | - | 98,175 | |||||||||||||||||||||
| Stock-based compensation | - | - | - | - | 2,500 | - | 2,500 | |||||||||||||||||||||
| Net loss | - | - | - | - | - | (708,682 | ) | (708,682 | ) | |||||||||||||||||||
| Balance, September 30, 2021 | 37,500 | $ | 21,703 | 90,396,596 | $ | 90,397 | $ | 163,644,453 | $ | (173,181,441 | ) | $ | (9,424,888 | ) | ||||||||||||||
Nine-months and Three-months Ended September 30, 2020
| Series
B Convertible Preferred Stock | Common Stock | Additional | Total | |||||||||||||||||||||||||
| Par | Paid-in | Accumulated | Stockholders’ | |||||||||||||||||||||||||
| Shares | Amount | Shares | Value | Capital | Deficit | Deficiency | ||||||||||||||||||||||
| Balance, December 31, 2019 | 37,500 | $ | 21,703 | 417,507 | $ | 418 | $ | 159,042,145 | $ | (166,509,085 | ) | $ | (7,444,819 | ) | ||||||||||||||
| Issuances of common stock | - | - | 2,951,878 | 2,952 | 937,166 | - | 940,118 | |||||||||||||||||||||
| Net loss for the three months ended March 31, 2020 | - | - | - | - | - | (946,718 | ) | (946,718 | ) | |||||||||||||||||||
| Balance at March 31, 2020 | 37,500 | $ | 21,703 | 3,369,385 | $ | 3,370 | $ | 159,979,311 | $ | (167,455,803 | $ | (7,451,419 | ) | |||||||||||||||
| Issuances of common stock | - | - | 18,861,353 | 18,861 | 311,947 | - | 330,808 | |||||||||||||||||||||
| Note discounts | - | - | - | - | 90,000 | - | 90,000 | |||||||||||||||||||||
| Net loss | - | - | - | - | - | (816,137 | ) | (816,137 | ) | |||||||||||||||||||
| Balance, June 30, 2020 | 37,500 | $ | 21,703 | 22,230,738 | $ | 22,231 | $ | 160,381,258 | $ | (168,271,940 | ) | $ | (7,846,748 | ) | ||||||||||||||
| Issuances of common stock (after issuance and full conversion of Series H Preferred Stock) | - | - | 25,377,426 | 25,377 | 1,664,307 | - | 1,689,684 | |||||||||||||||||||||
| Note payable conversions | - | - | 1,355,080 | 1,355 | 9,379 | - | 10,734 | |||||||||||||||||||||
| Option grants | - | - | - | - | 337,500 | - | 337,500 | |||||||||||||||||||||
| Warrant exercises | - | - | 8,820,956 | 8,821 | (8,821 | ) | - | - | ||||||||||||||||||||
| Net loss | - | - | - | - | - | (1,479,355 | ) | (1,479,355 | ) | |||||||||||||||||||
| Balance, September 30, 2020 | 37,500 | $ | 21,703 | 57,784,200 | $ | 57,784 | $ | 162,383,623 | $ | (169,751,295 | ) | $ | (7,288,185 | ) | ||||||||||||||
See accompanying notes to condensed consolidated financial statements (unaudited).
| F-43 |
RESPIRERX PHARMACEUTICALS INC.
AND SUBSIDIARY
CONDENSED CONSOLIDATED STATEMENTS OF CASH FLOWS
(Unaudited)
| Nine-months Ended | ||||||||
| September 30, | ||||||||
| 2021 | 2020 | |||||||
| Cash flows from operating activities: | ||||||||
| Net loss | $ | (2,371,145 | ) | $ | (3,242,210 | ) | ||
| Adjustments to reconcile net loss to net cash used in operating activities: | ||||||||
| Amortization of original issue discount | 40,795 | 21,332 | ||||||
| Amortization of capitalized note costs and debt discounts | 300,510 | 280,183 | ||||||
| Stock-based compensation included in - | ||||||||
| General and administrative expenses | 28,000 | 315,000 | ||||||
| Research and development expenses | 26,250 | 22,500 | ||||||
| Gain on warrant exchange | (1,099 | ) | - | |||||
| Loss on extinguishment of debt | - | 389,902 | ||||||
| Foreign currency transaction (gain) loss | (70,220 | ) | (7,151 | ) | ||||
| Changes in operating assets and liabilities: | ||||||||
| (Increase) decrease in - | ||||||||
| Prepaid expenses | (29,796 | ) | (27,385 | ) | ||||
| Deferred financing cost | (167,976 | ) | - | |||||
| Increase (decrease) in - | ||||||||
| Accounts payable and accrued expenses | 470,280 | 1,062,163 | ||||||
| Accrued compensation and related expenses | 867,950 | 700,540 | ||||||
| Accrued interest payable | 105,829 | 134,402 | ||||||
| Net cash used in operating activities | (800,622 | ) | (350,724 | ) | ||||
| Cash flows from financing activities: | ||||||||
| Proceeds from convertible note borrowings | 754,500 | 274,750 | ||||||
| Capitalized note costs and original issue discount | (109,950 | ) | - | |||||
| Proceeds from common stock issuance | 117,299 | - | ||||||
| Proceeds from issuance of note payable to officer | - | 59,500 | ||||||
| Officer advance | 7,130 | - | ||||||
| Repayment of short-term notes payable | (35,078 | ) | - | |||||
| Borrowings on short-term notes payable, net of repayments | 66,452 | - | ||||||
| Net cash provided by financing activities | 800,353 | 334,250 | ||||||
| Cash and cash equivalents: | ||||||||
| Net increase/(decrease) | (269 | ) | (16,474 | ) | ||||
| Balance at beginning of period | 825 | 16,690 | ||||||
| Balance at end of period | $ | 556 | $ | 216 | ||||
(Continued)
| F-44 |
RESPIRERX PHARMACEUTICALS INC.
AND SUBSIDIARY
CONDENSED CONSOLIDATED STATEMENTS OF CASH FLOWS
(Unaudited)
(Continued)
Nine-months
Ended | ||||||||
| 2021 | 2020 | |||||||
| Supplemental disclosures of cash flow information: | ||||||||
| Cash paid for - | ||||||||
| Interest | $ | 6,653 | $ | 1,498 | ||||
| Income taxes | $ | - | $ | - | ||||
| Non-cash financing activities: | ||||||||
Deferred offering costs applied against equity proceeds | $ | 52,609 | $ | - | ||||
| Conversion of accounts payable to long-term liabilities | $ | 332,000 | $ | - | ||||
| Commitment shares issued with debt financing | $ | 58,500 | $ | - | ||||
| Shares issued with conversion of debt | $ | 235,885 | $ | - | ||||
| Debt discounts established for convertible debt | $ | 580,725 | $ | 90,000 | ||||
| Issuance of common stock for accrued compensation and benefits | $ | - | $ | 306,000 | ||||
| Increase in principal amount of convertible note | $ | 5,000 | $ | - | ||||
| Issuance of warrants as deemed dividend associated with most-favored nation provisions of convertible notes | $ | 378,042 | $ | - | ||||
| Debt and accrued interest converted to common stock | $ | 4,000 | $ | 950,241 | ||||
| Cashless warrant exercises | $ | 900 | $ | 15,638 | ||||
See accompanying notes to condensed consolidated financial statements (unaudited).
| F-45 |
RESPIRERX PHARMACEUTICALS INC.
AND SUBSIDIARY
NOTES TO CONDENSED CONSOLIDATED FINANCIAL STATEMENTS
(Unaudited)
1. Organization and Basis of Presentation
Organization
RespireRx Pharmaceuticals Inc. (“RespireRx”) was formed in 1987 under the name Cortex Pharmaceuticals, Inc. to engage in the discovery, development and commercialization of innovative pharmaceuticals for the treatment of neurological and psychiatric disorders. On December 16, 2015, RespireRx filed a Certificate of Amendment to its Second Restated Certificate of Incorporation (as amended, the “Certificate of Incorporation”) with the Secretary of State of the State of Delaware to amend its Second Restated Certificate of Incorporation to change its name from Cortex Pharmaceuticals, Inc. to RespireRx Pharmaceuticals Inc. In August 2012, RespireRx acquired Pier Pharmaceuticals, Inc. (“Pier”), which is now a wholly owned subsidiary. Pier was a clinical stage biopharmaceutical company developing a pharmacologic treatment for obstructive sleep apnea (“OSA”) and had been engaged in research and clinical development activities which activities are now in RespireRx.
Basis of Presentation
The condensed consolidated financial statements are of RespireRx and its wholly-owned subsidiary, Pier (collectively referred to herein as the “Company,” “we” or “our,” unless the context indicates otherwise). The condensed consolidated financial statements of the Company at September 30, 2021 and for the three-months and nine-months ended September 30, 2021 and 2020, are unaudited. In the opinion of management, all adjustments (including normal recurring adjustments) have been made that are necessary to present fairly the condensed consolidated financial position of the Company, the condensed results of operations, condensed changes in stockholders’ deficiency and condensed changes in cash flows as of and for the periods ended September 30, 2021and 2020. Condensed consolidated operating results for the interim periods presented are not necessarily indicative of the results to be expected for a full fiscal year. The consolidated balance sheet at December 31, 2020 has been derived from the Company’s audited consolidated financial statements at such date. For comparative purposes, certain 2021 and 2020 amounts, including, but not limited to, share and per share amounts, par value and additional paid-in capital have been adjusted to a post-reverse stock split basis which occurred on January 5, 2021.
The condensed consolidated financial statements and related notes have been prepared pursuant to the rules and regulations of the Securities and Exchange Commission (the “SEC”). Accordingly, certain information and note disclosures normally included in financial statements prepared in accordance with United States generally accepted accounting principles (“GAAP”) have been omitted pursuant to such rules and regulations. These condensed consolidated financial statements should be read in conjunction with the consolidated financial statements and other information included in the Company’s Annual Report on Form 10-K for the fiscal year ended December 31, 2020 as filed with the SEC on April 15, 2021 (“2020 Form 10-K”).
2. Business
The mission of the Company is to develop innovative and revolutionary treatments to combat disorders caused by disruption of neuronal signaling. We are developing treatment options that address conditions that affect millions of people, but for which there are limited or poor treatment options, including obstructive sleep apnea (“OSA”), attention deficit hyperactivity disorder (“ADHD”), epilepsy, chronic pain, including inflammatory and neuropathic pain, recovery from spinal cord injury (“SCI”), as well as other areas of interest based on results of preclinical and clinical studies to date.
RespireRx is developing a pipeline of new drug products based on our broad patent portfolios across two distinct drug platforms:
| (i) | our pharmaceutical cannabinoids platform (which we refer to as ResolutionRx) is developing compounds that target the body’s endocannabinoid system, and in particular, the re-purposing of dronabinol, an endocannabinoid CB1 and CB2 receptor agonist, for the treatment of OSA. Dronabinol is already approved by the FDA for other indications. | |
| (ii) | our neuromodulators platform (which we refer to as EndeavourRx) is made up of two programs: (a) our AMPAkines program, which is developing proprietary compounds that are positive allosteric modulators (“PAMs”) of AMPA-type glutamate receptors to promote neuronal function and (b) our GABAkines program, which is developing proprietary compounds that are PAMs of GABAA receptors, and which was established pursuant to our entry into a patent license agreement (the “UWMRF Patent License Agreement”) with the University of Wisconsin-Milwaukee Research Foundation, Inc., an affiliate of the University of Wisconsin-Milwaukee (“UWMRF”). |
Financing our Platforms
Our major challenge has been to raise substantial equity or equity-linked financing to support research and development plans for our pharmaceutical cannabinoid and neuromodulator platforms, while minimizing the dilutive effect to pre-existing stockholders. At present, we believe that we are hindered primarily by our public corporate structure, our common stock not being listed on a national exchange, and low market capitalization as a result of our low stock price. Our shares are quoted on the OTCQB, the OTC Market’s venture market.
For this reason, the Company has implemented an internal restructuring plan through which our two drug platforms have been reorganized into separate business units and may in the future be organized into subsidiaries of RespireRx. We believe that by creating one or more subsidiaries to further the aims of ResolutionRx and EndeavourRx, it may be possible, through separate finance channels, to optimize the asset values of each. We are also planning to commence, assuming the SEC qualifies the offering, a securities offering pursuant to Regulation A under the Securities Act. See Note 9. Subsequent Events – Filing of Form 1-A for further information.
| F-46 |
Going Concern
The Company’s condensed consolidated financial statements have been presented on the basis that it is a going concern, which contemplates the realization of assets and satisfaction of liabilities in the normal course of business. The Company has incurred net losses of $708,682 and $2,371,145 for the three-months and nine-months ended September 30, 2021, respectively, and $4,301,211 for the fiscal year ended December 31, 2020, as well as negative operating cash flows of $800,622 for the nine-months ended September 30, 2021 and $513,001 for the fiscal year ended December 31, 2020. The Company also had a stockholders’ deficiency of $9,424,888 at September 30, 2021 and expects to continue to incur net losses and negative operating cash flows for the foreseeable future. As a result, management has concluded that there is substantial doubt about the Company’s ability to continue as a going concern, and the Company’s independent registered public accounting firm, in its report on the Company’s consolidated financial statements for the year ended December 31, 2020, expressed substantial doubt about the Company’s ability to continue as a going concern.
The Company is currently, and has for some time, been in significant financial distress. It has extremely limited cash resources and current assets and has no ongoing source of sustainable revenue. Management is continuing to address various aspects of the Company’s operations and obligations, including, without limitation, debt obligations, financing requirements, intellectual property, licensing agreements, legal and patent matters and regulatory compliance, and has taken steps to continue to raise new debt and equity capital to fund the Company’s business activities from both related and unrelated parties.
The Company is continuing its efforts to raise additional capital in order to be able to pay its liabilities and fund its business activities on a going forward basis, including the pursuit of the Company’s planned research and development activities. The Company regularly evaluates various measures to satisfy the Company’s liquidity needs, including development and other agreements with collaborative partners and, when necessary, seeking to exchange or restructure the Company’s outstanding securities. The Company is evaluating certain changes to its operations and structure to facilitate raising capital from sources that may be interested in financing only discrete aspects of the Company’s development programs. Such changes could include a significant reorganization, which may include the formation of one or more subsidiaries into which one or more programs may be contributed. As a result of the Company’s current financial situation, the Company has limited access to external sources of debt and equity financing. Accordingly, there can be no assurance that the Company will be able to secure additional financing in the amounts necessary to fully fund its operating and debt service requirements. If the Company is unable to access sufficient cash resources, the Company may be forced to discontinue its operations entirely and liquidate.
3. Summary of Significant Accounting Policies
Principles of Consolidation
The accompanying condensed consolidated financial statements are prepared in accordance with GAAP and include the financial statements of RespireRx and its wholly-owned subsidiary, Pier. Intercompany balances and transactions have been eliminated in consolidation.
Use of Estimates
The preparation of financial statements in conformity with GAAP requires management to make estimates and assumptions. These estimates and assumptions affect the reported amounts of assets and liabilities, disclosure of contingent assets and liabilities at the date of the financial statements and the reported amounts of revenues and expenses during the reporting period. Significant estimates include, among other things, accounting for potential liabilities, and the assumptions used in valuing stock-based compensation issued for services. Actual amounts may differ from those estimates.
Reverse Stock Split on January 5, 2021
On January 5, 2021, the Company effected a ten to one reverse-stock split of its common stock. Every ten shares of the “old” common stock was exchanged for one “new” share of common stock rounded down to the nearest whole share with any fractional shares of common stock paid to the stockholder in cash. Option and warrant issuances prior to January 5, 2021 have also been proportionately adjusted by dividing the number of shares into which such options and warrants may exercise by ten and multiplying the exercise price by ten. The effect of the reverse-stock split has been reflected retroactively in the Company’s consolidated financial statements as of December 31, 2020 and any interim periods in 2020. Certain amount with respect to 2019 that appear in these condensed consolidated financial statements have also been reflected on a post reverse-stock split basis.
| F-47 |
Concentration of Credit Risk
Financial instruments that potentially subject the Company to concentrations of credit risk consist primarily of cash and cash equivalents. The Company limits its exposure to credit risk by investing its cash with high quality financial institutions. The Company’s cash balances may periodically exceed federally insured limits. The Company has not experienced a loss in such accounts to date.
Value of Financial Instruments
The authoritative guidance with respect to value of financial instruments established a value hierarchy that prioritizes the inputs to valuation techniques used to measure value into three levels and requires that assets and liabilities carried at value be classified and disclosed in one of three categories, as presented below. Disclosure as to transfers into and out of Levels 1 and 2, and activity in Level 3 value measurements, is also required.
Level 1. Observable inputs such as quoted prices in active markets for an identical asset or liability that the Company has the ability to access as of the measurement date. Financial assets and liabilities utilizing Level 1 inputs include active-exchange traded securities and exchange-based derivatives.
Level 2. Inputs, other than quoted prices included within Level 1, which are directly observable for the asset or liability or indirectly observable through corroboration with observable market data. Financial assets and liabilities utilizing Level 2 inputs include fixed income securities, non-exchange based derivatives, mutual funds, and fair-value hedges.
Level 3. Unobservable inputs in which there is little or no market data for the asset or liability which requires the reporting entity to develop its own assumptions. Financial assets and liabilities utilizing Level 3 inputs include infrequently-traded, non-exchange-based derivatives and commingled investment funds, and are measured using present value pricing models.
The Company determines the level in the value hierarchy within which each value measurement falls in its entirety, based on the lowest level input that is significant to the value measurement in its entirety. In determining the appropriate levels, the Company performs an analysis of the assets and liabilities at each reporting period end.
The carrying amounts of financial instruments (consisting of cash, cash equivalents, and accounts payable and accrued expenses) are considered by the Company to be representative of the respective values of these instruments due to the short-term nature of those instruments. With respect to the note payable to SY Corporation Co., Ltd. (“SY Corporation”) and the convertible notes payable, management does not believe that the credit markets have materially changed for these types of borrowings since the original borrowing date. The Company considers the carrying amounts of the notes payable to officers, inclusive of accrued interest, to be representative of the respective values of such instruments due to the short-term nature of those instruments and their terms.
Deferred Financing Costs
Costs incurred in connection with ongoing debt and equity financings, including legal and accounting fees, are deferred until the related financing is either completed or abandoned.
Costs related to abandoned debt or equity financings are charged to operations in the period of abandonment. Costs related to completed equity financings are netted against the proceeds.
Debt Issuance Costs
The Company presents debt issuance costs related to debt obligations in its consolidated balance sheet as a direct deduction from the carrying amount of that debt obligation, consistent with the presentation for debt discounts.
| F-48 |
Convertible Notes Payable
Convertible notes are evaluated to determine if they should be recorded at amortized cost. To the extent that there are associated warrants, commitment shares of common stock or a beneficial conversion feature, the convertible notes and equity or equity-linked securities are evaluated to determine if there are embedded derivatives to be identified, bifurcated and valued in connection with and at the time of such financing.
Extinguishment of Debt and Settlement of Liabilities
The Company accounts for the extinguishment of debt and settlement of liabilities by comparing the carrying value of the debt or liability to the value of consideration paid or assets given up and recognizing a loss or gain in the condensed consolidated statement of operations in the amount of the difference in the period in which such transaction occurs. See Note 4 for additional information.
Prepaid Insurance
Prepaid insurance represents the premiums paid in March 2021 for directors and officers insurance and other insurance in April 2021. The amounts of prepaid insurance amortizable in the ensuing twelve-month period are recorded as prepaid insurance in the Company’s consolidated balance sheet at each reporting date and then amortized to the Company’s consolidated statement of operations for each reporting period.
Stock-Based Awards
The Company periodically issues common stock and stock options to officers, directors, Scientific Advisory Board members, consultants and vendors for services rendered. Such issuances vest and expire according to terms established at the issuance date of each grant.
The Company accounts for stock-based payments to officers, directors, outside consultants and vendors by measuring the cost of services provided in exchange for equity awards based on the grant date fair value of the awards, with the cost recognized as compensation expense on the straight-line basis in the Company’s consolidated financial statements over the vesting period of the awards.
Stock grants and stock options, which are sometimes subject to time-based vesting, are measured at the grant date fair value and charged to operations ratably over the vesting period.
The value of stock options granted as stock-based payments is determined utilizing the Black-Scholes option-pricing model, and is affected by several variables, the most significant of which are the life of the equity award, the exercise price of the stock option as compared to the fair market value of the common stock on the grant date, and the estimated volatility of the common stock over the term of the equity award. Estimated volatility is based on the historical volatility of the Company’s common stock. The risk-free interest rate is based on the U.S. Treasury yield curve in effect at the time of grant. The fair market value of common stock is determined by reference to the quoted market price of the Company’s common stock.
Stock and stock option grants and warrants issued to non-employees as compensation for services to be provided to the Company is accounted for based upon the fair value of the services provided or the estimated fair value of the stock option or warrant, whichever can be more clearly determined. Management uses the Black-Scholes option-pricing model to determine the fair value of the stock options and warrants issued by the Company. The Company recognizes this expense over the period in which the services are provided.
Stock and stock option grants and warrants issued to non-employees for services already rendered represent settlement of debt or liabilities and are accounted for by valuing both the amount of the amount of debt or liabilities settled and the value of the stock, stock option or warrant grants and recording a gain or loss as appropriate.
Stock and stock option grants and warrants issued to officers, directors, employees and affiliates in settlement of accrued compensation or other amounts payable are recorded at the amount of the liability settled. Therefore, there are no gains or losses recorded with respect to such settlements.
| F-49 |
There were no stock or stock option grants during the nine-months ended September 30, 2021.
The Company recognizes the amortized value of stock-based payments in general and administrative costs and in research and development costs, as appropriate, in the Company’s condensed consolidated statements of operations. The Company issues new shares of common stock to satisfy stock option and warrant exercises. There were no stock options exercised during the nine-months ended September 30, 2021 and 2020, respectively.
There were warrants to purchase 380,568 shares of common stock issued as compensation or for services during the nine-months ended September 30, 2021 and none during the nine-months ended September 30, 2020. Warrants, if issued for services, are typically issued to placement agents or brokers for fund raising services. In addition warrants to purchase 31,766,883 shares of common stock were issued to lenders during the nine-months ended September 30, 2021 with respect to or in association with the issuance of convertible notes to such lenders. Warrant are not issued from any of the Company’s stock and option plans, from which options issued to non-employees for services are typically issued.
Income Taxes
The Company accounts for income taxes under an asset and liability approach for financial accounting and reporting for income taxes. Accordingly, the Company recognizes deferred tax assets and liabilities for the expected impact of differences between the financial statements and the tax basis of assets and liabilities.
The Company records a valuation allowance to reduce its deferred tax assets to the amount that is more likely than not to be realized. In the event the Company was to determine that it would be able to realize its deferred tax assets in the future in excess of its recorded amount, an adjustment to the deferred tax assets would be credited to operations in the period such determination was made. Likewise, should the Company determine that it would not be able to realize all or part of its deferred tax assets in the future, an adjustment to the deferred tax assets would be charged to operations in the period such determination was made.
Pursuant to Internal Revenue Code Sections 382 and 383, use of the Company’s net operating loss and credit carryforwards may be limited if a cumulative change in ownership of more than 50% occurs within any three-year period since the last ownership change. The Company may have had a change in control under these Sections. However, the Company does not anticipate performing a complete analysis of the limitation on the annual use of the net operating loss and tax credit carryforwards until the time that it anticipates it will be able to utilize these tax attributes.
As of September 30, 2021, the Company did not have any unrecognized tax benefits related to various federal and state income tax matters and does not anticipate any material amount of unrecognized tax benefits within the next 12 months.
The Company is subject to U.S. federal income taxes and income taxes of various state tax jurisdictions. As the Company’s net operating losses have yet to be utilized, all previous tax years remain open to examination by Federal authorities and other jurisdictions in which the Company currently operates or has operated in the past.
The Company accounts for uncertainties in income tax law under a comprehensive model for the financial statement recognition, measurement, presentation and disclosure of uncertain tax positions taken or expected to be taken in income tax returns as prescribed by GAAP. The tax effects of a position are recognized only if it is “more-likely-than-not” to be sustained by the taxing authority as of the reporting date. If the tax position is not considered “more-likely-than-not” to be sustained, then no benefits of the position are recognized. As of September 30, 2021, the Company had not recorded any liability for uncertain tax positions. In subsequent periods, any interest and penalties related to uncertain tax positions will be recognized as a component of income tax expense.
Foreign Currency Transactions
The note payable to SY Corporation, which is denominated in a foreign currency (the South Korean Won), is translated into the Company’s functional currency (the United States Dollar) at the exchange rate on the balance sheet date. The foreign currency exchange gain or loss resulting from translation is recognized in the related condensed consolidated statements of operations.
| F-50 |
Research and Development
Research and development costs include compensation paid to management directing the Company’s research and development activities, including but not limited to compensation paid to our Chief Scientific Officer who is also our Executive Chairman, and fees paid to consultants and outside service providers and organizations (including research institutes at universities), and other expenses relating to the acquisition, design, development and clinical testing of the Company’s treatments and product candidates.
License Agreements
Obligations incurred with respect to mandatory payments provided for in license agreements are recognized ratably over the appropriate term, as specified in the underlying license agreement, and are recorded as liabilities in the Company’s condensed consolidated balance sheet, with a corresponding charge to research and development costs in the Company’s condensed consolidated statement of operations. Obligations incurred with respect to milestone payments provided for in license agreements are recognized when it is probable that such milestone will be reached and are recorded as liabilities in the Company’s condensed consolidated balance sheet, with a corresponding charge to research and development expenses in the Company’s condensed consolidated statement of operations.
Patent Costs
Due to the significant uncertainty associated with the successful development of one or more commercially viable products based on the Company’s research efforts and any related patent applications, all patent costs, including patent-related legal and filing fees, are expensed as incurred and recorded as general and administrative expenses.
Earnings per Share
The Company’s computation of earnings per share (“EPS”) includes basic and diluted EPS. Basic EPS is measured as the income (loss) attributable to common stockholders divided by the weighted average common shares outstanding for the period. Diluted EPS is similar to basic EPS but presents the dilutive effect on a per share basis of potential common shares (e.g., warrants and options) as if they had been converted at the beginning of the periods presented, or issuance date, if later. Potential common shares that have an anti-dilutive effect (i.e., those that increase income per share or decrease loss per share) are excluded from the calculation of diluted EPS.
Net loss attributable to common stockholders consists of net loss, as adjusted for actual and deemed preferred stock dividends declared, amortized or accumulated.
Loss per common share is computed by dividing net loss by the weighted average number of shares of common stock outstanding during the respective periods. Basic and diluted loss per common share is the same for all periods presented because all warrants and stock options outstanding are anti-dilutive.
At September 30, 2021 and 2020 the Company excluded the outstanding shares of common stock in the table below, into which the securities described below may convert, thereby entitling the holders thereof to acquire shares of common stock, from the Company’s calculation of earnings per share, as the effect would have been anti-dilutive.
| September 30, | ||||||||
| 2021 | 2020 | |||||||
| Series B convertible preferred stock | 1 | 1 | ||||||
| Convertible notes payable | 40,542,856 | 4,723,986 | ||||||
| Common stock warrants | 59,505,140 | 28,809,358 | ||||||
| Common stock options | 7,111,924 | 7,166,094 | ||||||
| Total | 107,159,921 | 40,699,439 | ||||||
Reclassifications
Certain comparative figures in 2020 have been reclassified to conform to the current quarter’s presentation. These reclassifications were immaterial, both individually and in the aggregate.
| F-51 |
Recent Accounting Pronouncements
In August 2020, the FASB issued Accounting Standards Update No. 2020-06, Debt – Debt with Conversion and Other Options (Subtopic 470-20) and Derivatives and Hedging—Contracts in Entity’s Own Equity (Subtopic 815-40). The subtitle is Accounting for Convertible Instruments and Contracts in an Entity’s Own Equity. This Accounting Standard Update (“ASU”) addresses complex financial instruments that have characteristics of both debt and equity. The application of this ASU would reduce the number of accounting models for convertible debt instruments and convertible preferred stock. Limiting the accounting models would result in fewer embedded conversion features being separately recognized from the host contract as compared with current GAAP. Convertible instruments that continue to be subject to separation models are (1) those with embedded conversion features that are not clearly and closely related to the host contract, that meet the definition of a derivative, and that do not qualify for a scope exception from derivative accounting and (2) convertible debt instruments issued with substantial premiums for which the premiums are recorded as paid-in capital. The Company has historically issued complex financial instruments and has considered whether embedded conversion features have existed within those contracts or whether derivatives would appropriately be bifurcated. To date, no such bifurcation has been necessary. However, it is possible that this ASU may have a substantial impact on the Company’s financial statements. Management is evaluating the potential impact. This ASU becomes effective for fiscal years beginning after December 15, 2023.
In January 2020, the FASB issued ASU 2020-01, Clarifying the Interactions between Topic 321, Topic 323, Equity Method and Joint Ventures, and Topic 815, Derivatives and Hedging which represents an amendment clarifying the interaction between accounting standards related to equity securities, equity method investments and certain derivatives. The guidance is effective for fiscal years beginning after December 15, 2020 and interim periods within those years. Management has evaluated the guidance and determined that the implementation of such guidance does not have a material impact on our condensed consolidated financial statements.
4. Notes Payable
Convertible Notes Payable
On April 1, 2021, May 3, 2021, May 10, 2021, June 30, 2021 and August 31, 2021, the Company closed on financings, pursuant to which, five convertible notes were issued to four separate investors, due in each case, one year from the effective date (which for the each closing was March 31, 2021, April 30, 2021, May 10, 2021, June 29, 2021 and August 31, 2021, respectively), with maturity amounts of $112,500, $150,000, $150,000, $115,000 and $115,000, respectively. In addition, the noteholders received as consideration, warrants to purchase 2,400,000, 3,200,000, 3,200,000, 2,453,333 and 5,750,000 shares of common stock, respectively, each exercisable for a period of five years at an exercise price of $0.02 per share. The August 31, 2021 issuance triggered the most-favored nation provisions of four notes already outstanding, resulting in the issuance of 15,121,667 additional warrants on September 7, 2021. As a result of the note issuances, the Company received net proceeds of $96,750, $123,400, $123,400, $100,000 and $103,500 respectively, for an aggregate of $547,050. The difference between the maturity amounts and the net proceeds were due to original issue discounts, in four cases, investor legal fees and in two cases, broker fees. The five notes are convertible at a fixed price of $0.02 per share and bear interest at 10% per year which interest is guaranteed regardless of prepayment. The Company has the right to prepay the notes during the first six months subject to prepayment premiums that range from 0% to 15% (100% to 115% of the maturity amount plus accrued interest and any default interest and similar costs).
The Company periodically issues convertible notes with similar characteristics. As described in the table below, as of September 30, 2021, there were nine such notes outstanding (including the convertible notes described in the paragraph above), two of which were satisfied in full by conversion of both principal and interest and one of which was satisfied in part, principal only, during that period. These notes all have or had a fixed conversion price of $0.02 per share of common stock, subject to adjustment in certain circumstances. All notes but one had an annual interest rate of 10% which was guaranteed in full. One note had an annual interest rate of 8%. The convertible notes had an original issue discount (“OID”), debt issuance costs (“DIC”) that were capitalized by the Company, a warrant (“WT”) or commitment shares (“CS”) and in three cases a beneficial conversion feature (“BCF”). The OID, CN, WTs, CSs and BCF allocated values are amortized over the life of the notes to interest expense. All notes mature or matured nine to fifteen months from their issuance date. All notes were prepayable by the Company during the first six months, subject to prepayment premiums that range from 100% to 115% of the maturity amount plus accrued interest. If not earlier paid, the notes were convertible by the holder into the Company’s common stock. Two of the notes were paid before maturity.
| F-52 |
The table below summarizes the convertible notes with similar characteristics outstanding as of September 30, 2021 and the repayments by conversion during the nine-months ended September 30, 2021:
| Inception Date | Maturity date | Original Principal Amount | Interest rate | Original aggregate DIC, OID, Wts, CS and BCF | Cumulative amortization of DIC, OID, Wts, CS and BCF | Accrued coupon interest | Repayment
by conversion | Balance
sheet carrying amount at September 30, 2021 inclusive of accrued interest | ||||||||||||||||||||||
| July 2, 2020 | April 2, 2021 | $ | 137,500 | 10.00 | % | $ | (44,423 | ) | $ | 44,423 | $ | 6,875 | $ | (144,375 | ) | $ | - | |||||||||||||
| July 28, 2020 | July 28, 2021 | 45,000 | 8.00 | % | (5,000 | ) | — | 2,743 | (25,000 | ) | 17,743 | |||||||||||||||||||
| July 30, 2020 | October 30, 2021 | 75,000 | 10.00 | % | (27,778 | ) | 27,778 | 4,136 | (79,136 | ) | - | |||||||||||||||||||
| February 17, 2021 | November 17, 2021 | 112,000 | 10.00 | % | (112,000 | ) | 91,608 | 9,161 | - | 100,769 | ||||||||||||||||||||
| April 1, 2021 | March 31, 2022 | 112,500 | 10.00 | % | (112,500 | ) | 56,404 | 5,640 | - | 62,044 | ||||||||||||||||||||
| May 3, 2021 | April 30, 2022 | 150,000 | 10.00 | % | (150,000 | ) | 62,877 | 6,288 | - | 69,165 | ||||||||||||||||||||
| May 10, 2021 | May 10, 2022 | 150,000 | 10.00 | % | (150,000 | ) | 58,767 | 5,877 | - | 64,644 | ||||||||||||||||||||
| June 30, 2021 | June 29, 2022 | 115,000 | 10.00 | % | (115,000 | ) | 29,301 | 2,930 | - | 32,231 | ||||||||||||||||||||
| August 31, 2021 | August 31, 2022 | 115,000 | 10.00 | % | (109,675 | ) | 9,015 | 945 | - | 15,285 | ||||||||||||||||||||
| Total | $ | 1,012,000 | $ | (826,376 | ) | $ | 380,173 | $ | 44,595 | $ | (248,511 | ) | $ | 361,881 | ||||||||||||||||
In addition to what appears in the table above, there is outstanding accrued interest of $2,747 from a prior floating rate convertible note that has not been paid in cash or by conversion as of September 30, 2021.
On July 27, 2021, the maturity date of the note scheduled to mature on July 28, 2021, was extended to December 1, 2021 and the original and remaining principal amount of the note was increased by $5,000 from $40,000 to $45,000 and from $15,000 to $20,000 respectively, with interest on the incremental increase in principal amount accruing from the note inception date. Since the Company did not receive any cash proceeds from the increase in the principal amount of the note, the increase was recorded as a debt discount to be amortized to interest expense through the maturity date.
In addition to the convertible notes with similar characteristics described above, on December 31, 2018 and January 2, 2019, the Company issued convertible notes to a single investor totaling $35,000 of maturity amount with accrued interest of $10,399 as of September 30, 2021. The number of shares of common stock (or preferred stock) into which these notes may convert is not determinable. The warrants to purchase 19,000 shares of common stock issued in connection with the sale of these notes and other convertible notes issued December 2018 and March 2019 are exercisable at a fixed price of $15.00 per share of common stock, provide no right to receive a cash payment, and included no reset rights or other protections based on subsequent equity transactions, equity-linked transactions or other events and expire on December 30, 2023.
Other convertible notes were also sold to investors in 2014 and 2015 (“Original Convertible Notes”), which aggregated a total of $579,500, and had a fixed interest rate of 10% per annum. The Original Convertible Notes have no reset rights or other protections based on subsequent equity transactions, equity-linked transactions or other events. The warrants to purchase shares of common stock issued in connection with the sale of the Original Convertible Notes have either been exchanged as part of note and warrant exchange agreements executed in April and May of 2016 or expired on September 15, 2016.
| F-53 |
The remaining outstanding Original Convertible Notes (including those for which default notices have been received) consist of the following at September 30, 2021 and December 31, 2020:
| September 30, 2021 | December 31, 2020 | |||||||
| Principal amount of notes payable | $ | 75,000 | $ | 75,000 | ||||
| Accrued interest payable | 74,763 | 64,357 | ||||||
| $ | 149,256 | $ | 139,357 | |||||
As of September 30, 2021, principal and accrued interest on the Original Convertible Note that is subject to a default notice accrues annual interest at 12% instead of 10%, totaled $52,337, of which $27,337 was accrued interest. As of December 31, 2020, principal and accrued interest on Original Convertible Notes subject to default notices totaled $48,700 of which $23,700 was accrued interest.
As of September 30, 2021 all of the outstanding Original Convertible Notes, inclusive of accrued interest, were convertible into an aggregate of 1,317 shares of the Company’s common stock. Such Original Convertible Notes will continue to accrue interest until exchanged, paid or otherwise discharged. There can be no assurance that any of the additional holders of the remaining Original Convertible Notes will exchange their Original Convertible Notes.
Note Payable to SY Corporation Co., Ltd.
On June 25, 2012, the Company borrowed 465,000,000 Won (the currency of South Korea, equivalent to approximately $400,000 United States Dollars as of that date) from and executed a secured note payable to SY Corporation Co., Ltd., (“SY Corporation”). The note accrues simple interest at the rate of 12% per annum and had a maturity date of June 25, 2013. The Company has not made any payments on the promissory note. At June 30, 2013 and subsequently, the promissory note was outstanding and in default, although SY Corporation has not issued a notice of default or a demand for repayment. Management believes that SY Corporation is in default of its obligations under its January 2012 license agreement, as amended, with the Company, but the Company has not yet issued a notice of default. The Company has in the past made several efforts towards a comprehensive resolution of the aforementioned matters involving SY Corporation. During the nine-months ended September 30, 2021, there were no further communications between the Company and SY Corporation.
The promissory note is secured by collateral that represents a lien on certain patents owned by the Company, dating back to January, August and September 2007, including composition of matter patents for certain of the Company’s high impact ampakine compounds and the low impact ampakine compounds CX2007 and CX2076, and other related compounds that the Company is no longer developing and where patent rights date back to January, August and September 2007. The security interest does not extend to the Company’s patents for its low impact ampakine compounds, such as CX717, CX1739 and CX1942 or certain related method of use patents.
The note payable to SY Corporation consists of the following at September 30, 2021 and December 31, 2020:
| September 30, 2021 | December 31, 2020 | |||||||
| Principal amount of note payable | $ | 399,774 | $ | 399,774 | ||||
| Accrued interest payable | 447,266 | 411,384 | ||||||
| Foreign currency transaction adjustment | (16,838 | ) | 53,393 | |||||
| $ | 830,202 | $ | 864,551 | |||||
Interest expense with respect to this promissory note was $35,881 and $36,013 for the nine-months ended September 30, 2021 and 2020, respectively, and $12,092 for the three-months ended September 30, 2021 and 2020.
Notes Payable to Officers and Former Officers
The following amounts were charged to interest expense with respect to notes payable to Dr. Arnold S. Lippa: $3,096 and $2,848 for the three-months ended September 30, 2021 and 2020, respectively, and $ 9,193 and $8,481 for the nine-months ended September 30, 2021 and 2020, respectively.
The following amounts were charged to interest expense with respect to notes payable to Dr. James S. Manuso: $4,702 and $4,274 for the three-months ended September 30, 2021 and 2020, respectively, and $13,954 and $12,714 for the nine-months ended September 30, 2021 and 2020, respectively.
| F-54 |
Other Short-Term Notes Payable
Other short-term notes payable at September 30, 2021 and December 31, 2020 consisted of premium financing agreements with respect to various insurance policies. At September 30, 2021, a premium financing agreement was payable in the initial amount of $81,672 (after payment of a deposit of $20,347), with interest at 11% per annum, in eight monthly installments of $10,635. In addition, there is $9,238 of short-term financing of office and clinical trials insurance premiums. At September 30, 2021 and December 31, 2020, the aggregate amount of the short-term notes payable was $35,982 and $4,608 respectively.
5. Settlement and Payment Agreements
On February 21, 2020, Sharp Clinical Services, Inc. (“Sharp”), a vendor of the Company, filed a complaint against the Company in the Superior Court of New Jersey Law Division, Bergen County related to a December 16, 2019 demand for payment of past due invoices inclusive of late fees totaling $103,890 of which $3,631 related to late fees, seeking $100,259 plus 1.5% interest per month on outstanding unpaid invoices. On May 29, 2020, a default was entered against the Company, and on September 4, 2020, a final judgment by default was entered against the Company in the amount of $104,217. On March 3, 2021, we executed a settlement agreement with Sharp (the “Sharp Settlement Agreement”), and on March 9, 2021, Sharp requested of the Bergen (NJ) County Sheriff, the return of the Writ of Execution which resulted in a release of the lien in favor of Sharp. The Sharp Settlement Agreement calls for a payment schedule of ten $10,000 payments due on April 1, 2021 and every other month thereafter, and permitted early settlement at $75,000 if the Company had paid Sharp that lower total by August 1, 2021, but the Company did not pay Sharp that lower amount by that date. The Company has recorded a liability to Sharp of $63,859 as of September 30, 2021 after payments totaling $30,000 pursuant to the Sharp Settlement Agreement.
By letter dated February 5, 2016, the Company received a demand from a law firm representing Salamandra, LLC (“Salamandra”) alleging an amount due and owing for unpaid services rendered. On January 18, 2017, following an arbitration proceeding, an arbitrator awarded Salamandra the full amount sought in arbitration of $146,082. Additionally, the arbitrator granted Salamandra attorneys’ fees and costs of $47,937. All such amounts have been accrued as of September 30, 2021 and December 31, 2020, including accrued interest at 4.5% annually from February 26, 2018, the date of the judgment, through September 30, 2021, totaling $29,897. The Company had previously entered into a settlement agreement with Salamandra that is no longer in effect. The Company has approached Salamandra seeking to negotiate a new settlement agreement. A lien with respect to the amounts owed is in effect.
On February 23, 2021, our bank received two New Jersey Superior Court Levies totaling $320,911 related to amounts owed to Sharp and Salamandra which amounts were not in dispute. The bank debited our accounts and restricted access to those accounts pursuant to the liens placed on the accounts. Our accounts were debited for $1,559 on February 23, 2021, which represented all of the cash in our accounts on that date.
On April 29, 2021, the Company entered into a payment and settlement agreement with the University of California Innovation and Entrepreneurship, pursuant to which it agreed to a payment schedule that is reflected in accounts payable and accrued expenses in the Company’s condensed consolidated financial statements. The total amount due is $234,657. The agreed payment schedule is for the Company to pay $10,000 on each of July 1, 2021, September 1, 2021, November 1, 2021, January 1, 2022 and March 31, 2022. If the Company pays an aggregate of $175,000 on or before March 31, 2022, the amounts will be considered paid in full with no further amounts due. If an aggregate of $175,000 has not been paid by March 31, 2022, the remaining unpaid amount up to an aggregate of the original amount of $234,657 would be due and payable. The payments due on July 1, 2021 and September 1, 2021 were timely paid.
On September 14, 2021, the Company and DNA Healthlink, Inc. (“DNA Healthlink”) entered into a settlement agreement (the “ DNA Healthlink Settlement Agreement”) regarding $410,000 in unpaid accounts payable owed by the Company to DNA Healthlink (the “DNA Healthlink Settlement Amount”) for services provided by DNA Healthlink to the Company pursuant to an agreement by and between the Company and DNA Healthlink dated October 15, 2014. Under the terms of the DNA Healthlink Settlement Agreement, the Company is obligated to pay to DNA Healthlink the full DNA Healthlink Settlement Amount as follows: twelve monthly payments of $8,000each commencing on November 15, 2021, followed by twelve monthly payments of $10,000 each commencing on November 15, 2022, followed by twelve monthly payments of $15,000 each commencing on November 15, 2023, followed by one final payment of $14,000 on November 15, 2024. If, prior to March 14, 2023, the Company receives one or more upfront license fee payments or any other similar fee or fees from one or more strategic partners that aggregate at least fifteen million dollars ($15,000,000.00) (“Upfront Fees”), then the full DNA Healthlink Settlement Amount, less any amounts previously paid, will be accelerated and become due and payable in full within ninety (90) days of receipt of any Upfront Fees. As a result of the DNA Healthlink Settlement Agreement, the Company recorded a gain with respect to vendor settlements of $62,548.
An annual obligation payable to the University of Illinois of $100,000 that was originally due on December 31, 2020 pursuant to the 2014 License Agreement was extended to April 19, 2021 and was paid in full on April 1, 2021.
By email dated July 21, 2016, the Company received a demand from an investment banking consulting firm that represented the Company in 2012 in conjunction with the Pier transaction alleging that $225,000 is due and payable for investment banking services rendered. Such amount has been included in accrued expenses at September 30, 2021 and December 31, 2020. See Note 1 for additional information on the Pier transaction.
The Company is periodically the subject of various pending and threatened legal actions and claims. In the opinion of management of the Company, adequate provision has been made in the Company’s consolidated financial statements as of September 30, 2021 and December 31, 2020 with respect to such matters, including, specifically, the matters noted above. The Company intends to vigorously defend itself if any of the matters described above results in the filing of a lawsuit or formal claim.
| F-55 |
6. Stockholders’ Deficiency
Preferred Stock
RespireRx has authorized a total of 5,000,000 shares of preferred stock, par value $0.001 per share. As of September 30, 2021 and December 31, 2020, 37,500 shares were designated as Series B Convertible Preferred Stock (non-voting, “Series B Preferred Stock”).
Series B Preferred Stock outstanding as of September 30, 2021 and 2020 consisted of 37,500 shares issued in a May 1991 private placement. The shares of Series B Preferred Stock are convertible into 1 share of common stock. RespireRx may redeem the Series B Preferred Stock for $25,001 at any time upon 30 days prior notice.
Although other series of preferred stock have been designated, no other shares of preferred stock are outstanding. As of September 30, 2021 and December 31, 2020, 3,504,424 shares of preferred stock were undesignated and may be issued with such rights and powers as the Board of Directors may designate.
Common Stock
RespireRx has authorized 2,000,000,000 (2 billion) shares of common stock, par value $0.001 per share. There are 90,396,596 shares of the Company’s common stock outstanding as of September 30, 2021. After reserving for conversions of convertible debt and convertible preferred stock, as well as exercises of common stock purchase options (granted and available for grant within the 2014 and 2015 stock and stock option plans) and warrants and the issuance of Pier contingent shares and before accounting for incremental contract excess reserves, there were 1,786,678,967 shares of the Company’s common stock available for future issuances as of September 30, 2021. After accounting for incremental excess reserves contractually required by the various convertible notes and certain warrants, there were 1,702,729,267 shares of common stock available for future issuances as of September 30, 2021. On May 27, 2021, a holder of a warrant exercised the warrant in part, exercising into 900,000 shares of common stock on a cashless basis, what would have exercised into 1,665,958 shares of common stock on a cash basis. The Company did not receive any cash proceeds from the exercise. No other warrants were exercised during the nine-months ended September 30, 2021. No options were exercised during the nine-months ended September 30, 2021. No warrants or options were exercised after September 30, 2021. See Note 9 for a description of a partial warrant exercise and a conversion of a portion of the principal amount of one convertible note, both occurring on November 8, 2021.
Common Stock Warrants
A summary of warrant activity for the nine-months ended September 30, 2021 is presented below.
| Number
of Shares | Weighted Average Exercise Price | Weighted
Average Remaining Contractual Life (in Years) | ||||||||||
| Warrants outstanding at December 31, 2020 | 28,809,352 | $ | 0.1528 | 2.64 | ||||||||
| Issued | 33,432,841 | 0.0200 | ||||||||||
| Expired | (8,595 | ) | ||||||||||
| Cancelled upon exchange | (1,062,500 | ) | 0.0700 | |||||||||
| Exercised - cashless | (1,665,958 | ) | 0.0200 | |||||||||
| Warrants outstanding at September 30, 2021 | 59,505,140 | $ | 0.0721 | 2.12 | ||||||||
| Warrants exercisable at September 30, 2020 | 28,809,358 | $ | 0.1474 | 2.89 | ||||||||
| Warrants exercisable at September 30, 2021 | 59,505,140 | $ | 0.0721 | 2.12 | ||||||||
The exercise prices of common stock warrants outstanding and exercisable are as follows at September 30, 2021:
| Exercise Price | Warrants Outstanding and Exercisable (Shares) | Expiration
Date | ||||||
| $ | 0.016 | 2,212,500 | May 17, 2022 | |||||
| $ | 0.020 | 31,386,315 | March 31, 2026-September 7, 2021 | |||||
| $ | 0.039 | 208,227 | May 10, 2026 | |||||
| $ | 0.047 | 172,341 | May 3, 2026 | |||||
| $ | 0.070 | 25,377,426 | September 30, 2023 | |||||
| $ | 11.00 -27.50 | 148,331 | December 31, 2021-December 30, 2023 | |||||
| 59,505,140 | ||||||||
| F-56 |
Based on a value of $0.0230 per share on September 30, 2021, there were 33,598,815 exercisable in-the-money common stock warrants as of September 30, 2021.
A summary of warrant activity for the nine months ended September 30, 2020 is presented below.
| Number
of Shares | Weighted Average Exercise Price | Weighted
Average Remaining Contractual Life (in Years) | ||||||||||
| Warrants outstanding at December 31, 2019 | 219,104 | $ | 18.7109 | 3.44 | ||||||||
| Issued including issuances as a result of anti-dilution protections | 39,585,039 | 0.0521 | - | |||||||||
| Expired | (25,435 | ) | (59.9808 | ) | - | |||||||
| Exercised | (10,969,350 | ) | (0.0161 | ) | - | |||||||
| Warrants outstanding and exercisable at September 30, 2020 | 28,809,358 | $ | 0.1474 | 2.89 | ||||||||
The exercise prices of common stock warrants outstanding and exercisable at September 30, 2020 ranged from $0.0161 to $79.30 and these warrants were exercisable into an aggregate of 28,809,358 shares, which warrants have expired or will expire as applicable, between February 28, 2021 and October 22, 2024.
The exercise prices of common stock warrants outstanding and exercisable at September 30, 2020 ranged from $0.016 to $79.30 and these warrants were exercisable into an aggregate of 28,809,358 shares, which warrants have expired or will expire, as applicable, between February 28, 2021 and October 22, 2024.
Based on a value of $0.054 per share on September 30, 2020, there were 28,652,426 exercisable in-the-money common stock warrants as of that date.
Stock Options
On March 18, 2014, the stockholders of RespireRx holding a majority of the votes to be cast on the issue approved the adoption of RespireRx’s 2014 Equity, Equity-Linked and Equity Derivative Incentive Plan (the “2014 Plan”), which had been previously adopted by the Board of Directors, subject to stockholder approval. The Plan permits the grant of options and restricted stock in addition to stock appreciation rights and phantom stock, to directors, officers, employees, consultants and other service providers of the Company.
On June 30, 2015, the Board of Directors adopted the 2015 Stock and Stock Option Plan (as amended, the “2015 Plan”). As of September 30, 2021, there are 15,757,542 shares available in the 2015 Plan. On July 29, 2021, the Company amended the 2015 Plan to increase the number of shares by an additional 7,000,000 to 22,898,526 shares. The Company has not and does not intend to present the 2015 Plan to stockholders for approval.
Information with respect to the Black-Scholes variables used in connection with the evaluation of the fair value of stock-based compensation costs and fees is provided at Note 3.
A summary of stock option activity for the six-months ended September 30, 2021 is presented below.
Number of Shares | Weighted Average Exercise Price | Weighted Average Remaining Contractual Life (in Years) | ||||||||||
| Options outstanding at December 31, 2020 | 7,165,215 | $ | 1.96 | 4.98 | ||||||||
| Expired | (53,291 | ) | 73.78 | - | ||||||||
| Options outstanding at September 30, 2021 | 7,111,924 | $ | 1.43 | 3.88 | ||||||||
| Options exercisable at September 30, 2021 | 7,111,924 | $ | 1.43 | 3.88 | ||||||||
| F-57 |
The exercise prices of common stock options outstanding and exercisable were as follows at September 30, 2021:
| Exercise Price | Options Outstanding (Shares) | Options Exercisable (Shares) | Expiration Date | |||||||||
| $ | 0.0540 | 1,700,000 | 1,700,000 | September 30, 2025 | ||||||||
| $ | 0.072 | 5,050,000 | 5,050,000 | July 31, 2025 | ||||||||
| $ | 7.00-$195.00 | 361,924 | 361,924 | September 12, 2021 - December 9, 2027 | ||||||||
| 7,111,924 | 7,111,924 | |||||||||||
There was no deferred compensation expense for the outstanding and unvested stock options at September 30, 2021.
Based on a fair value of $0.023 per share on September 30, 2021, there were no exercisable in-the-money common stock options as of that date.
Reserved and Unreserved Shares of Common Stock
As of September 30, 2021, there are 2,000,000,000 shares of common stock authorized, of which 90,396,596 are issued and outstanding. As of September 30, 2021, there are outstanding options to purchase 7,111,924 shares of common stock and 6,325 and 15,757,542 shares available for issuance under the 2014 Plan and 2015 Plan, respectively. There are 649 Pier contingent shares of common stock that may be issued under certain circumstances. As of September 30, 2021, there are 40,542,856 shares issuable upon conversion of convertible notes. As of June 30, 2021, there are 59,505,150 shares that may be issued upon exercise of outstanding warrants. As of September 30, 2021, the Series B Preferred Stock may convert into 1 share of common stock. Therefore, the Company is reserving 146,542,854 shares of common stock for future issuances with respect to conversions and exercises as well as for the Pier contingent shares. In addition, certain convertible notes and related warrants impose an additional contractual reserve requirement, above the number of shares into which such convertible notes and related warrants may convert or exercise respectively. Although the Company does not anticipate having to issue such shares, such incremental additional contractual reserves total 83,949,701 shares of common stock.
7. Related Party Transactions
Dr. Arnold S. Lippa and Jeff E. Margolis, officers and directors of RespireRx since March 22, 2013, have indirect ownership and managing membership interests in Aurora Capital LLC (“Aurora”) through interests held in its members, and Jeff E. Margolis is also an officer of Aurora. Aurora was a boutique investment banking firm specializing in the life sciences sector that ceased its securities related activities in April 2021. On May 5, 2021, Aurora filed to withdraw its membership with FINRA and its registration with the SEC which withdrawal became effective in July 2021. Although Aurora has not provided services to RespireRx during the nine-months ended September 30, 2021 or the fiscal year ended December 31, 2020, Aurora had previously provided services to the Company and there remains $96,000 owed to Aurora by RespireRx which amount is included in accounts payable and accrued expenses as of September 30, 2021.
A description of advances and notes payable to officers is provided at Note 4.
8. Commitments and Contingencies
Pending or Threatened Legal Action and Claims
The Company is periodically the subject of various pending and threatened legal actions and claims. In the opinion of management of the Company, adequate provision has been made in the Company’s condensed consolidated financial statements as of September 30, 2021 and 2020 with respect to such matters. See Note 5 for additional items and details.
| F-58 |
Significant Agreements and Contracts
The Company entered into a consulting contract with David Dickason effective September 15, 2020 pursuant to which Mr. Dickason was appointed and serves as the Company’s Senior Vice President of Pre-Clinical Product Development on an at-will basis at the rate of $250 per hour. The Company recorded but did not pay cash compensation expense pursuant to this agreement of $72,375 for the nine-months ended September 30, 2021.
See Note 5 for details of other agreements and contracts.
Employment Agreements
Timothy L. Jones, Arnold S. Lippa and Jeff E. Margolis have similar employment agreements. Mr. Jones was appointed as RespireRx’s President and Chief Executive Officer on May 6, 2020. Dr. Lippa is RespireRx’s Chief Scientific Officer and Executive Chairman and Mr. Margolis is the Company’s Senior Vice President, Chief Financial Officer, Treasurer and Secretary. Dr. Lippa’s and Mr. Margolis’ employment agreements became effective on August 18, 2015. All three agreements are subject to automatic annual extensions on September 30th of each year beginning with the initial termination date if not earlier terminated, subject to notice in accordance with the terms of the agreements. Mr. Jones’ initial termination date is September 30, 2023 and Dr. Lippa’s and Mr. Margolis’ agreements are in their automatic extension periods.
The table below summarized the current cash commitments to each individual through the next September 30th renewal date and in the case of Mr. Jones, through September 30, 2023.
| Contract year ending | Contract year ending | |||||||||||||||||||||||||||||||
| September 30, 2022 | September 30, 2023 | |||||||||||||||||||||||||||||||
| Twelve months | Twelve months | |||||||||||||||||||||||||||||||
| Base | Guaranteed | Base | Guaranteed | |||||||||||||||||||||||||||||
| Salary | Benefits | Bonus | Total | Salary | Benefits | Bonus | Total | |||||||||||||||||||||||||
| Timothy L. Jones | $ | 265,500 | $ | 39,600 | $ | 300,000 | $ | 605,100 | $ | 300,000 | $ | 39,600 | $ | 300,000 | $ | 639,600 | ||||||||||||||||
| Arnold S. Lippa | 300,000 | 39,600 | — | 339,600 | — | — | — | — | ||||||||||||||||||||||||
| Jeff E. Margolis | 300,000 | 21,600 | — | 321,600 | — | — | — | — | ||||||||||||||||||||||||
| $ | 865,500 | $ | 100,800 | $ | 300,000 | $ | 1,266,300 | $ | 300,000 | $ | 39,600 | $ | 300,000 | $ | 639,600 | |||||||||||||||||
Under certain circumstances base salaries may be contractually increased or the executives may become eligible for additional benefits and base salaries may be increased at the discretion of the Board of Directors. All executives are eligible for stock and stock option and similar grants at the discretion of the Board or Directors.
The payment of certain amounts reflected in the table above have been voluntarily deferred indefinitely and payments against accrued compensation may be made based upon the Company’s ability to make such payments.
The second amendment to the employment agreement of Timothy Jones, the Company’s President and Chief Executive Officer (the “Employment Agreement”) became effective on September 27, 2021 (“Amendment No. 2”). Mr. Jones is also a director of the Company.
Amendment No. 2 reduces Mr. Jones’ annual Base Salary from $300,000 to $231,000 from October 1, 2021 through March 31, 2022 and restores the annual Base Salary to $300,000 on April 1, 2022. Each month commencing on October 1, 2021 and ending on the last day of the month and on a monthly accumulating basis thereafter until March 31, 2022, during which the Company raises at least $200,000 up to an aggregate amount equal to or greater than $1,200,000, the Base Salary must be paid in cash at the rate of $19,250 per month. In addition, the $150,000 guaranteed bonus that would have been due on September 30, 2021 was adjusted to $0 in accordance with the terms of Amendment No. 2.
After March 31, 2022, until such time as at least $2,500,000 has been raised, Mr. Jones’ salary may be accrued and remain unpaid, at the discretion of the Board. In the event that a payment due in accordance with the Amendment No. 2 payment schedule, is not timely paid, and is not cured by payment during the subsequent month inclusive of such subsequent month’s payment due, Mr. Jones’s Base Salary will revert back to $300,000 per year, the payment schedule becomes null and void and the adjustment to a $0 Guaranteed Bonus will also be deemed null and void and a payment of $150,000 will become due and payable as of the same date that the Base Salary is adjusted to $300,000.
If $10,000,000 is raised by April 30, 2022, Mr. Jones’ annual Base Salary will be adjusted to $375,000. Furthermore, if the Board determines that a sufficient amount of funds have been raised or is otherwise available to fund the Company’s operations on an ongoing basis, some or all of the accrued and unpaid Base Salary or Guaranteed Bonus as described in Amendment No. 2 may be paid in cash.
UWMRF Patent License Agreement
On August 1, 2020, the (“Effective Date”), the Company and UWMRF executed the UWMRF Patent License Agreement pursuant to which, the Company has an exclusive license to commercialize GABAkine products based on UWMRF’s rights in certain patents and patent applications, and a non-exclusive license to commercialize products based on UWMRF’s rights in certain technology that is not the subject of the patents or patent applications. UWMRF maintains the right to use, and, upon the approval of the Company, to license, these patent and technology rights for any non-commercial purpose, including research and education. The UWMRF Patent License Agreement expires upon the later of the expiration of the Company’s payment obligations to UWMRF or the expiration of the last remaining licensed patent granted thereunder, subject to early termination upon the occurrence of certain events. The License Agreement also contains a standard indemnification provision in favor of UWMRF and confidentiality provisions obligating both parties.
| F-59 |
Under the UWMRF Patent License Agreement, in consideration for the licenses granted, the Company will pay to UWMRF the following: (i) patent filing and prosecution costs incurred by UWMRF prior to the effective date, paid in yearly installments over three years from the Effective Date; (ii) annual maintenance fees, beginning on the second anniversary of the Effective Date, which annual maintenance fees terminate upon the Company’s payment of royalties pursuant to clause (iv) below; (iii) milestone payments, paid upon the occurrence of certain dosing events of patients during clinical trials and certain approvals by the FDA; and (iv) royalties on net sales of products developed with the licenses, subject to minimum annual payments and to royalty rate adjustments based on whether separate royalty payments by the Company yield an aggregate rate beyond a stated threshold. The Company has also granted UWMRF certain stock appreciation rights with respect to the Company’s neuromodulator programs, subject to certain limitations, and will pay to UWMRF certain percentages of revenues generated from sublicenses of the licenses provided under the UWMRF Patent License Agreement by the Company to third parties.
University of Wisconsin-Milwaukee Outreach Services Agreement
On July 12, 2021, the Company and the Board of Regents of the University of Wisconsin System on behalf of the University of Wisconsin-Milwaukee (“UWM”) entered into an Outreach Services Agreement pursuant to which UWM agreed to provide, among other molecules, multiple milligram to gram quantities of KRM-II-81 (GABAkine) and the Company agreed to pay UWM an annual sum of $75,000 payable in three installments of $25,000 each beginning October 12, 2021, which amount was timely paid, and on a quarterly basis thereafter. The agreement terminates on June 30, 2022 unless extended upon consent of both parties.
University of Illinois 2014 Exclusive License Agreement
The Company and the University of Illinois entered into the Exclusive License Agreement (the “2014 License Agreement”) effective September 18, 2014, pursuant to which the Company obtained (i) exclusive rights to several issued and pending patents in numerous jurisdictions and (ii) the non-exclusive right to certain technical information that is generated by the University of Illinois in connection with certain clinical trials as specified in the 2014 License Agreement, all of which relate to the use of cannabinoids for the treatment of sleep related breathing disorders. The Company is developing dronabinol (Δ9-tetrahydrocannabinol), a cannabinoid, for the treatment of OSA, the most common form of sleep apnea.
The 2014 License Agreement provides for various commercialization and reporting requirements that commenced on June 30, 2015. In addition, the 2014 License Agreement provides for various royalty payments, including a royalty on net sales of 4%, payment on sub-licensee revenues of 12.5%, and a minimum annual royalty beginning in 2015 of $100,000, which is due and payable on December 31 of each year beginning on December 31, 2015. The minimum annual royalty obligation of $100,000 due on December 31, 2020, was extended to April 19, 2021 and was paid in full on April 1, 2021. One-time milestone payments may become due based upon the achievement of certain development milestones. $75,000 will be due within 5 days of any one of the following, (a) dosing of the first patient with a dronabinol product in a Phase 2 human clinical study anywhere in the world that is not sponsored by the University of Illinois, (b) dosing of the first patient in a Phase 2 human clinical study anywhere in the world with a low dose dronabinol (defined as less than or equal to 1 mg), or (c) dosing of the first patient in a Phase 1 human clinical study anywhere in the world with a proprietary reformulation of dronabinol. $350,000 will be due within five days after the dosing of the first patient is a Phase III human clinical trial anywhere in the world. $500,000 will be due within five days after the first NDA filing with FDA or a foreign equivalent. $1,000,000 will be due within twelve months of the first commercial sale. One-time royalty payments may also become due and payable. Annual royalty payments may also become due. In the year after the first application for market approval is submitted to the FDA or a foreign equivalent and until approval is obtained, the minimum annual royalty will increase to $150,000. In the year after the first market approval is obtained from the FDA or a foreign equivalent and until the first sale of a product, the minimum annual royalty will increase to $200,000. In the year after the first commercial sale of a product, the minimum annual royalty will increase to $250,000.
The Company recorded charges to operations of $25,000 during the three-months ended September 30, 2021 and 2020, respectively, and $75,000 during the nine-months ended September 30, 2021 and 2020, respectively, with respect to its minimum annual royalty obligation, which is included in research and development expenses in the Company’s condensed consolidated statement of operations for the three-months and nine-months ended September 30, 2021 and 2020. As discussed above, the Company did not pay the amount due on December 31, 2020 for which the Company was granted an extension until April 19, 2021 and which was paid in full on April 1, 2021.
| F-60 |
Noramco Inc. - Dronabinol Development and Supply Agreement
On September 4, 2018, RespireRx entered into a dronabinol Development and Supply Agreement with Noramco Inc., one of the world’s major dronabinol manufacturers, which Noramco subsequently assigned to its subsidiary, Purisys LLC (the “Purisys Agreement”). Under the terms of the Purisys Agreement, Purisys has agreed to (i) provide all of the active pharmaceutical ingredient (“API”) estimated to be needed for the clinical development process for both the first- and second-generation products (each a “Product” and collectively, the “Products”), three validation batches for New Drug Application (“NDA”) filing(s) and adequate supply for the initial inventory stocking for the wholesale and retail channels, subject to certain limitations, (ii) maintain or file valid drug master files (“DMFs”) with the FDA or any other regulatory authority and provide the Company with access or a right of reference letter entitling the Company to make continuing reference to the DMFs during the term of the agreement in connection with any regulatory filings made with the FDA by the Company, (iii) participate on a development committee, and (iv) make available its regulatory consultants, collaborate with any regulatory consulting firms engaged by the Company and participate in all FDA or Drug Enforcement Agency (“DEA”) meetings as appropriate and as related to the API. We now refer to the second-generation product as our proprietary formulation or proprietary product and have de-emphasized the first-generation product.
In consideration for these supplies and services, the Company has agreed to purchase exclusively from Purisys during the commercialization phase all API for its Products (as defined in the Development and Supply Agreement) at a pre-determined price subject to certain producer price index adjustments and agreed to Purisys’ participation in the economic success of the commercialized Product or Products up to the earlier of the achievement of a maximum dollar amount or the expiration of a period of time.
There was no activity during the three-months ended and nine-months ended September 30, 2021 or 2020 with respect to the Purisys Agreement.
Transactions with Bausch Health Companies Inc. (formerly known as Biovail Laboratories International SRL)
Beginning in March 2010, the Company entered into a series of asset purchase and license agreements with Biovail Laboratories International SRL which later merged with Valeant Pharmaceuticals International, Inc. which was later renamed Bausch Health Companies Inc. (“Bausch”).
In March 2011, the Company entered into a new agreement with Bausch to reacquire the ampakine compounds, patents and rights that Bausch had acquired from the Company in March 2010. The new agreement provided for potential future payments of up to $15,150,000 by the Company based upon the achievement of certain developments, including new drug application submissions and approval milestones pertaining to an intravenous dosage form of the ampakine compounds for respiratory depression, a therapeutic area not currently pursued by the Company. Bausch is also eligible to receive additional payments of up to $15,000,000 from the Company based upon the Company’s net sales of an intravenous dosage form of the compounds for respiratory depression.
At any time following the completion of Phase 1 clinical studies and prior to the end of Phase 2A clinical studies, Bausch retains an option to co-develop and co-market intravenous dosage forms of an ampakine compound as a treatment for respiratory depression and vaso-occlusive crises associated with sickle cell disease. In such an event, the Company would be reimbursed for certain development expenses to date and Bausch would share in all such future development costs with the Company. If Bausch makes the co-marketing election, the Company would owe no further milestone payments to Bausch and the Company would be eligible to receive a royalty on net sales of the compound by Bausch or its affiliates and licensees.
There was no activity during the three-months ended and nine-months ended September 30, 2021 or 2020 that affect the Bausch agreement.
Summary of Principal Cash Obligations and Commitments
The following table sets forth the Company’s principal cash obligations and commitments for the next five fiscal years as of September 30, 2021, aggregating $2,166,177. License agreement amounts included in the 2021 column represents amounts contractually due from October 1, 2021 through December 31, 2021 (three months) and in each of the subsequent years, represents the full year. Employment agreement amounts included in the 2021 column represent amounts contractually due from October 1, 2021 through September 30, 2022 (twelve months) and September 30, 2023 in the case of the employment agreement of Timothy Jones, when such contracts expire unless extended pursuant to the terms of the contracts.
| Payments Due By Year | ||||||||||||||||||||||||
| Total | 2021 | 2022 | 2023 | 2024 | 2025 | |||||||||||||||||||
| License agreements | $ | 520,277 | $ | 25,000 | $ | 120,092 | $ | 140,185 | $ | 110,000 | $ | 115,000 | ||||||||||||
| Research and development contracts | 50,000 | - | 50,000 | - | - | - | ||||||||||||||||||
| Employment agreements (1) | 1,605,900 | 232,950 | 1,118,250 | 254,700 | - | - | ||||||||||||||||||
| Total | $ | 2,166,177 | $ | 257,950 | $ | 1,288,342 | $ | 394,885 | $ | 110,000 | $ | 115,000 | ||||||||||||
| (1) | The payment of certain of such amounts has been deferred indefinitely, as described above in “Employment Agreements”. |
9. Subsequent Events
Amendment to Mr. Jones’ Employment Agreement
See Note 8. Commitments and Contingencies – Significant Agreements and Contracts – Employment Agreements for a description of Amendment No. 2 of Mr. Jones’ employment agreement.
Filing of Form 1-A
On October 12, 2021, the Company filed amendment number one to its Form 1-A which had previously been filed on August 9, 2021 with the SEC with respect to a contemplated Tier 2 offering under Regulation A. The Form 1-A and preliminary offering circular contained therein is subject to further amendment. No securities may be offered prior to qualification of the offering by the Securities and Exchange Commission or in any jurisdiction in which such offer, solicitation or sale of securities would be unlawful before registration or qualification under the laws of such jurisdiction.
| F-61 |
Convertible Note and Related Transactions
On October 7, 2021, the Company and Dariusz Nasiek and Sara Nasiek JTTEN (the “Nasieks”) entered into a Securities Purchase Agreement (the “Nasiek SPA”) pursuant to which the Nasieks provided a sum of $103,500 (the “Nasiek Consideration”) to the Company in return for a convertible promissory note (the “Nasiek Note”) with a face amount of $115,000 (which difference in value as compared to the Nasiek Consideration is due to an original issue discount of $11,500), and a common stock purchase warrant (the “Nasiek Warrant”) exercisable for five years at an exercise price of $0.02 per share on a cash or cashless basis, to purchase up to 5,750,000 shares of the Company’s common stock, par value $0.001. In addition, and to induce the Nasieks to enter into the Nasiek SPA, the Company and the Nasieks entered into a Piggy-Back Registration Rights Agreement (the “Nasiek Registration Rights Agreement”) under which the Company has agreed to provide certain piggy-back registration rights under the Securities Act of 1933, as amended with respect to the common stock issuable pursuant to the Nasiek SPA.
The Note obligates the Company to pay by October 7, 2022 (the “Nasiek Maturity Date”) a principal amount of $115,000 together with interest at a rate equal to 10% per annum. The first twelve months of interest, equal to $11,500, is guaranteed and earned in full as of the effective date. Any amount of principal or interest that is not paid by the Nasiek Maturity Date would bear interest at the rate of 24% from the Nasiek Maturity Date to the date it is paid.
The Nasieks have the right, in their discretion, at any time, to convert any outstanding and unpaid amount of the Nasiek Note into shares of common stock, provided that the conversion would not result in the Nasieks beneficially owning more than 4.99% of the Company’s then outstanding common stock. The Nasieks may convert at a per share conversion price equal to $0.02, subject to equitable adjustments for stock splits, stock dividends, combinations, recapitalizations, extraordinary distributions and similar events. Upon any conversion, all rights with respect to the portion of the Nasiek Note being so converted will terminate, except for the right to receive common stock or other securities, cash or other assets as provided for in the Nasiek Note.
The Company may, in the absence of an event of default, and with prior written notice to the Nasieks, prepay the outstanding principal amount under the Nasiek Note during the initial 180 day period after the effective date by making a payment to the Nasieks of an amount in cash equal to 115% of the outstanding principal, interest, default interest and other amounts owed. Under certain circumstances, including the occurrence of an event of default, a sale, merger or other business combination where the Company is not the survivor, or the conveyance or disposition of all or substantially all of the assets of the Company, the Company may be required to prepay in cash an amount equal to 125% of the outstanding principal, interest, default interest and other amounts owed. The Company’s wholly owned subsidiary, Pier Pharmaceuticals, Inc., provided an unlimited guarantee of the Company’s obligations under the Nasiek Note.
The Nasiek Note requires that the Company reserve the greater of (i) 8,625,000 shares of Common Stock or (ii) one and a half times the number of shares into which the Nasiek Note may convert. The Nasiek Warrant requires that the Company reserve three times the number of shares into which the Nasiek Warrant is at any time exercisable.
The Nasiek SPA includes, among other things: (1) the grant of an option to the Nasieks to incorporate into the Nasiek Note any terms applicable to a subsequent issuance of a convertible note or security by the Company that are more beneficial to an investor than the terms of the Nasiek SPA and Nasiek Note are to the Nasieks; and (2) certain registration rights by reference to the Nasiek Registration Rights Agreement, and the right to have any shares of common stock issued in connection with the conversion of the Nasiek Note or exercise of the Nasiek Warrant included in any Regulation A offering statement that the Company files with the Securities and Exchange Commission.
Partial Conversion of Convertible Note
On November 8, 2021, the Company received a notice of conversion with respect to a convertible note with a maturity date of November 17, 2021, pursuant to which the holder converted $80,000 of the $112,000 principal amount of the note into 4,000,000 shares of common stock which were issued by the Company. There remains $32,000 of principal amount plus accrued interest on that convertible note.
Cashless Warrant Exercise
On November 8, 2021, the Company received a partial cashless exercise notice with respect to a portion of a warrant which portion of such warrant would have been exercisable into 1,534,042 shares of common stock if exercised for cash. Pursuant to the cashless exercise, the Company issued 511,347 shares of common stock and cancelled a portion of the warrant with respect to 1,534,042 shares of common stock. The Company did not receive any proceeds from the cashless exercise.
| F-62 |
RESPIRERX PHARMACEUTICALS INC.
Best Efforts Offering of
$7,500,000 Maximum Offering Amount (375,000,000 Shares of Common Stock)

OFFERING CIRCULAR
December 13, 2021
Item 16. Index to Exhibits
| III-1 |
| III-2 |
| III-3 |
| III-4 |
| III-5 |
| III-6 |
| III-7 |
| * | Each of these Exhibits constitutes a management contract, compensatory plan or arrangement. |
| ** | Certain information has been omitted pursuant to Item 17 of Form 1-A because it is both not material and would be competitively harmful if publicly disclosed. When filing the document with its Current Report on Form 8-K, the Company undertook to furnish, supplementally, a copy of the unredacted exhibit to the Securities and Exchange Commission upon request. |
| III-8 |
SIGNATURES
Pursuant to the requirements of Regulation A, the issuer certifies that it has reasonable grounds to believe that it meets all of the requirements for filing on Form 1-A and has duly caused this offering statement to be signed on its behalf by the undersigned, thereunto duly authorized, in the City of Glen Rock, State of New Jersey, on the 13th day of December, 2021.
| RESPIRERX PHARMACEUTICALS INC. | ||
| By: | /s/ Timothy Jones | |
| Timothy Jones | ||
| President, Chief Executive Officer and Director | ||
Pursuant to the requirements of Regulation A, this Offering Statement has been signed by the following persons in the capacities and on the dates indicated.
| Signature | Title | Date | ||
| /s/ Timothy Jones | President, Chief Executive Officer and Director; Principal | December 13, 2021 | ||
| Timothy Jones | Executive Officer | |||
| /s/ Jeff E. Margolis | Senior Vice President, Chief Financial Officer, Secretary, | December 13, 2021 | ||
| Jeff E. Margolis | ||||
| /s/ Arnold S. Lippa Ph.D. | Chief Scientific Officer, Director and Executive Chairman | December 13, 2021 | ||
| Arnold S. Lippa Ph.D. | of the Board | |||
| Treasurer and Director; Principal Financial Officer and Principal Accounting Officer | ||||
| **** | Director | December 13, 2021 | ||
Kathryn MacFarlane |
**** By: |
/s/ Jeff E. Margolis | |
| Jeff E. Margolis | ||
| Attorney-in-Fact |
| III-9 |